TYPE 1 DIABETES MELLITUS
Epidemiology of Type 1 Diabetes
T1DM is one of the most common chronic diseases of childhood and is classified as an autoimmune disease. Most common autoimmune disorders predominantly affect females, but, T1DM equally affects males and females with a slight male predominance in younger children. This and other inconsistencies have raised questions as to whether T1DM is a “pure” auto-immune disease or whether the auto-immune component is a marker of a separate primary trigger (3,4). We discuss these issues later in this chapter.
The incidence and prevalence of T1DM vary by age, season, geographic location, and within different racial and ethnic groups. Of cases diagnosed before the age of 20, however, two peaks of T1DM presentation are observed; one between 5 and 7 years of age, and the other during puberty at the mid-teens (5). However, first presentation of T1DM actually is as common in adulthood as it is in childhood and is characterized by a milder course in adults; the term LADA, (Latent, Auto-immune, Diabetes of Adults) is used to describe this entity. A seasonal variation in the incidence of T1DM is also observed; the majority of new cases of T1DM are diagnosed mostly in autumn and winter (6). Findings from large T1DM registry studies such as the World Health Organization Multinational Project for Childhood Diabetes, known as the DIAMOND Project, EURODIAB and others monitor incidence and other epidemiological markers.
The World Health Organization Multinational Project for Childhood Diabetes, known as the DIAMOND Project (in 50 countries), EURODIAB (in Europe), and SEARCH for Diabetes in Youth (in the USA) were established to address the implications of diabetes in youth and describe the incidence of T1DM. Wide variations in incidence of T1DM exist throughout the world, lowest in China and Venezuela (0.1 per 100,000 per year) and highest in Finland and Sardinia (50-60 per 100,000 per year) (7). A multicenter study focusing on identifying the prevalence and incidence of diabetes by type, age, gender, and ethnicity found a 1.8% annual increase in the prevalence of T1DM among American youth from 2002-2003 to 2011-2012, whereas T2DM had increased 4.8% annually from 2002-2003 to 2011-2012 (Table 3) (8). The greatest increase was seen in youth of minority racial/ethnic groups (8). Similar rates of increase in T2DM in teens are reported from the UK, India, China and Japan.
Table 3.
Incidence of T1DM in the USA (per 100,000/year)
View in own window
| Age Group |
|---|
| 0-4 yr | 5-9 yr | 10-14 yr | 15-19 yr |
|---|
| Non-Hispanic White | 18.6 | 28.1 | 32.9 | 15.1 |
| African American | 9.7 | 16.2 | 19.2 | 11.1 |
| Hispanic American | 9.1 | 15.7 | 17.6 | 12.1 |
| American Indian | 4.1 | 5.5 | 7.1 | 4.8 |
| Asian and Pacific Islander American | 6.1 | 8.0 | 8.3 | 6.8 |
| All | 14.3 | 22.1 | 25.9 | 33.1 |
Although, there is a wide variance in the incidence and prevalence of diabetes throughout the world, the number of youths who are being diagnosed with T1DM has been growing at an annual rate of about 3 percent (9) and a similar increased annual rate was also observed among U.S. youth (10). This rising incidence of T1DM in children across the world in a short period of time clearly cannot be explained by genetic factors. Analytical epidemiological studies suggest that environmental risk factors, operating early in life, might be contributing to the increasing trend in incidence of T1DM (11,12).
On the basis of estimates for the number of people with diabetes in 2014, the cost of health care of diabetes in the US is estimated to be $105 billion per annum and the direct annual cost of diabetes in the world is $825 billion (13). However studies indicate that many more diabetic adults diagnosed as having T2DM phenotype actually have T1DM as defined by the presence of antibodies to islet cell components (14,15); the term LADA, Latent Autoimmune Diabetes of Adults, is often used to describe this group (16).
Natural History of Type 1 Diabetes
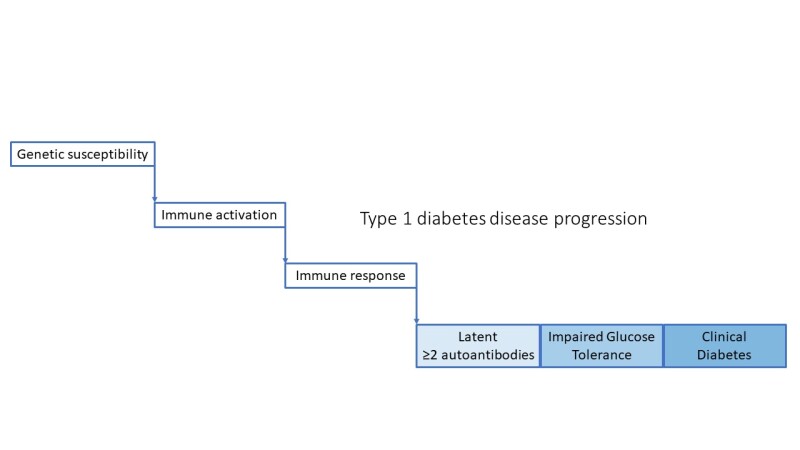
After immune activation in the setting of genetic susceptibility, the disease progresses through pre-symptomatic stages identified by presence of autoantibodies and impaired glucose intolerance, arising from further loss of β-cell function and ultimately resulting in clinical diabetes. ()
Structure and Functions of the Pancreas
Pancreatic ß cells secrete insulin and are found in the islets of Langerhans. These islets are specialized groups of a few hundred to a few thousand endocrine cells that are anatomically and functionally discrete from pancreatic exocrine tissue, the primary function of which is to secrete pancreatic enzymes into the duodenum. Normal subjects have about one million islets, which in total weigh only 1-2 grams and constitute less than 1% of the mass of the pancreas. Furthermore, islets are composed of various types of cells that are interconnected as a regulatory network to regulate the disposition of nutrients and their utilization for energy use and tissue growth and repair. At least 70% are ß cells localized in the core of the islets, surrounded by α-cells that secrete glucagon, δ-cells that secrete somatostatin, and PP cells that secrete pancreatic polypeptide. All the cells communicate with each other through their extracellular spaces and through gap junctions; communication is further modulated by a rich network of sympathetic and para sympathetic innervation.
Insulin, a peptide hormone composed of 51 amino acids is synthesized, packaged and secreted in pancreatic ß cells. Insulin is synthesized as preproinsulin in the ribosomes of rough endoplasmic reticulum. The preproinsulin is then cleaved to proinsulin that is transported to the Golgi apparatus where it is packaged into secretory granules. Most of the proinsulin is cleaved into equimolar amounts of insulin and connecting (or C)-peptide in the secretory granules. Because the C-peptide sequence differs from that of insulin, and because, unlike insulin, it is not extracted by the liver, it is possible to estimate β-cell insulin secretion by measuring C-peptide, even in the presence of insulin antibodies resulting from insulin replacement therapy that impair the ability to measure insulin directly. Similarly, because C-peptide is an index of endogenous insulin secretion, and because C-peptide is not extracted by the liver, the ratio of C-peptide: insulin should exceed 1; when it is less than 1, implying a high insulin value, exogenous insulin may have been used. This has diagnostic and forensic utility in diagnosing causes of hypoglycemia.
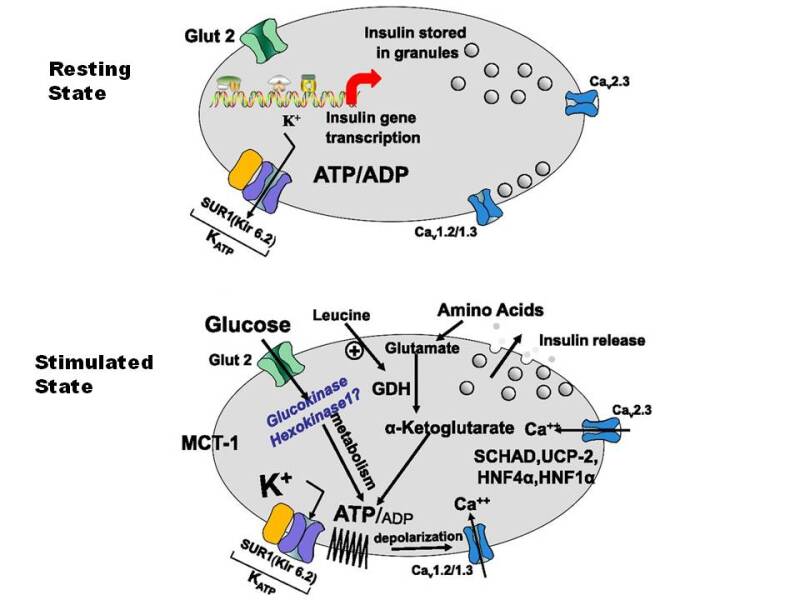
Glucose is a major regulator of insulin secretion (). When extracellular fluid glucose concentrations rise after a meal, glucose is taken up by the ß cells via glucose transporters, GLUT2 and GLUT1. Glucose is then phosphorylated into glucose-6-phosphate by islet specific glucokinase and metabolized, thereby increasing cellular ATP concentrations. The rise in ATP raises the resting ratio of ATP:ADP, that closes ATP dependent potassium channels (K-ATP) in the β-cell membrane, resulting in accumulation of intracellular potassium, causing membrane depolarization and influx of calcium via a voltage gated calcium channel. The rise in intracellular free calcium in ß-cells promotes margination of the secretory granules, their fusion with the cell membrane, and release of cell contents which include insulin into the extracellular space. An immediately releasable pool of insulin granules adjacent to the plasma membrane is responsible for an acute (first phase) insulin response; with ongoing stimulation, a pool of granules in the interior of the cell is mobilized and released as the “second phase” response. Amino acids also stimulate insulin release by a similar mechanism that involves the enzyme glutamate dehydrogenase which enables metabolism and ATP production by certain amino acids. Defects in the genes regulating these processes may result in diabetes if the K-ATP channel is prevented from closing normally (activating mutations) or syndromes of hyperinsulinemic hypoglycemia if the K-ATP channel is prevented from opening (inactivating mutations). These aspects are discussed in greater detail in the section on Monogenic forms of diabetes (see below).
Metabolic Derangements of Type 1 Diabetes
As the pancreatic ß cell mass declines in an islet cell antibody (ICA) positive person, the first metabolic abnormality discernable is a decline in the first phase of insulin release (FPIR) to an IVGTT (18). The insulin level after a 3-4 minute infusion of glucose at 0.5Gms/kg rises abruptly in normal children at about 8 years of age, perhaps coincident with the onset of adrenarche (19). In the relatives and children from the general population with positive ICA, a decline in the FPIR is a strong predictive marker of evolving diabetes (19-21).
Subsequently, in evolving T1DM there is a rise in the fasting glucose level followed by an inability to keep the two-hour, post-OGTT glucose level below 200mg/dl (11.1mM). Transient insulin resistance also occurs in untreated T1DM and is due to raised levels of free fatty acids (FFAs) from uncontrolled lipolysis (22), as well as decreased levels of hepatic glucokinase and insulin regulated GLUT 4 glucose transporters in adipocytes which contribute to the onset of symptomatic diabetes (23-25). Prolonged hyperglycemia itself likely impairs the ability to secrete insulin and when insulin replacement therapy begins, there is usually some recovery in the patient's ability to secrete insulin (the "honeymoon" period). However, within months to years, this partial recovery in endogenous insulin secretion ultimately fails. If it does not fail after 2 years, another form of diabetes, such as MODY should be suspected. Initially, the glucagon secreting cells within the pancreatic islets remain relatively preserved, resulting in excessive secretion of glucagon relative to insulin after protein meals (26). These elevated glucagon levels exacerbate the effects of the insulin deficiency, and promote lipolysis and ketogenesis, effects that can be partially reversed by an infusion of somatostatin (27). As the mass of islet cells decline, there is also loss of amylin, an islet cell hormone that down-regulates glucagon secretion. Thus, an analogue of amylin (pramlintide- marketed under the trade name Symlin) can be used as adjunctive therapy with insulin replacement. In time, with continued loss of islets, glucagon deficiency develops in established long standing T1DM, rendering patients more susceptible to insulin-induced hypoglycemia (26,28).
Insulin is the hormone of "feasting", promoting utilization and deposition of ingested nutrients into body stores, as well as having multiple anabolic effects in many tissues. Progressive insulin deficiency thus induces a starvation like state, associated with excessive hepatic and renal gluconeogenesis, decreased peripheral utilization of glucose, hyperglycemia with resultant glycosuria, loss of water and sodium salts, and proteolysis in muscle liberating amino acids such as alanine and glutamine as substrates for gluconeogenesis (29-31). Uncontrolled lipolysis leads to the rapid mobilization of fatty acids from adipose tissue and the increased delivery of fatty acids to the liver leading to the increased synthesis of triglycerides and secretion of very low-density lipoprotein (VLDL).
With severe insulin deficiency the fatty acids delivered to the liver are metabolized to yield beta hydroxybutyric and aceto-acetic acids (ketone bodies) and contribute to keto-acidosis. Ketoacidosis is a life-threatening metabolic decompensation that is characterized by hyperglycemia, dehydration, metabolic acidosis and ketosis, all the result of the effects of severe insulin deficiency as well as the counter-regulatory stress hormones, cortisol, growth hormone, catecholamines and glucagon. Specifically, hepatic glucokinase levels fall with insulinopenia, synthesis of hepatic triglyceride and glycogen levels decline, malonyl CoA falls and thereby carnitine palmitoyl transferase-I levels rise promoting the transport of fatty acyl-CoA into mitochondria with the formation of acetyl-CoA (32-34). In the liver, acetyl-CoA is converted into ß-hydroxybutyrate and acetoacetate in a proportion that depends upon the prevailing redox state, which provide an additional fuel substrates for muscle and brain (31,35,36). Lipoprotein lipases are also inactivated, leading to reduced hydrolysis of triglycerides that, if severe, may turn the serum milky with increased VLDL characteristic of the type 4 lipemic phenotype (37-39).
Genetic Susceptibility to Type 1 Diabetes
Individuals with autoimmune T1DM have inherited a number of quantitative trait loci (QTL) that encode protective and predisposing alleles which have exceeded the net genetic threshold required to predispose them to the disease (40). However, this genetic threshold (penetrance) is dependent in turn on chance interactions with greater predisposing than protective environmental forces. The multiple genetic influences in T1DM comprise a major effect from DR/DQ genotypes of the HLA complex (some 50% of the genetic effect), coupled to several other QTLs with minor influences (Table 4). All of the latter QTLs are not obligatory genetic elements themselves since they are of minor-influence, but they collectively interact to create additive influences on the genetic threshold. Siblings of a diabetic patient develop T1DM at about 15-fold greater frequency than persons in the general population (prevalence 1:250-300), vs. a value of 15. The HLA predisposition to T1DM is encoded by cis- and trans complementation DQA1*/DQB1* heterodimers which have an arginine at residue 52 of the A chain and a neutral amino acid (DQB1*0302, *0201) rather than a charged aspartic acid at residue 57 of the B chain (DQB1*0602/3 and DQB1*0301) (40), as modified by DRB1*04 subtypes (*0401 and *0405 are susceptible and *0403 and 6 are resistant types) (41) in the HLA genotype. Further, HLA-DP alleles have also been implicated, even though they are at a considerable recombination frequency away from the closely linked DR/DQ loci (42). Other genes involved include the variable number of tandem repeat (VNTR) alleles 5' to the insulin (INS) gene on chromosome 11p15, where the protective class III alleles (>200 repeats) are associated with increased expression of insulin in the thymus, leading to a more efficient eradication of insulin autoreactive T cells than class I alleles (26-63 repeats) that confer susceptibility to develop diabetes (43,44). There are also CTLA-4 gene polymorphisms on chromosome 2q that are associated with T1DM. CTLA-4 is an induced accessory molecule that is expressed on activated T cells. CTLA-4 interacts with B7.2 expressed by antigen presenting cells (APC), signaling apoptosis of T cells that become activated as part of an immune response, thereby confining the immune response. The non-obese diabetic (NOD) mouse, a model for autoimmune diabetes, has an enlarged lymphoid mass because of resistance of their T cells to undergo apoptosis, as do CTLA-4 knockout mice, which readily develop lymphocytic organ infiltrates like NOD mice. These genes thus collectively affect the general ability to be tolerant to "self" antigens. Another susceptibility locus, (the IDDM 4) in the genomic interval on chromosome 11q13harbors the high affinity IgE Fc receptor gene that has been linked to atopy and asthma, which are characterized byTh2 responses that may protect individuals against the development of anti- islet Th1 responses, and thereby protect against T1DM. There are other genomic intervals associated with or linked to T1DM that have been putatively mapped, but these mostly lack plausible candidate genes in the DNA region, and pathogenic mechanisms for them cannot yet be offered. The NOD mouse however has been subjected to extensive genetic mapping studies, in the hopes that genomic intervals harboring susceptibility or protective genes which are syntenic to humans will be discovered, thus hastening the identification of equivalent defective genes.
Table 4.
Genotypes of the HLA Complex Associated with Diabetes Mellitus
View in own window
| Locus | Chromosome | Candidate Genes/Microsatellites | References |
|---|
| IDDM1 | 6p21.3* | HLA-DQ/DR | (45,46) |
| IDDM2 | 11p15* | INS VNTR | (47,48) |
| IDDM3 | 15q26 | D15s107 | (49) |
| IDDM4 | 11q13 | MDU1, ZFM1, RT6, FADD/MORT1, LRP5 | (50,51) |
| IDDM5 | 6q24-27 | ESR, MnSOD | (52) |
| IDDM6 | 18q12-q21 | D18s487, D18s64, JK (Kidd locus) | (53) |
| IDDM7 | 2q31 | D2s152, IL-1, NEUROD, GALNT3 | (54) |
| IDDM8 | 6q25-27 | D6s264, D6s446, D6s281 | (52) |
| IDDM9 | 3q21-25 | D3s1303 | (55) |
| IDDM10 | 10p11-q11 | D10s193, D10s208, D10s588 | (56) |
| IDDM11 | 14q24.3-q31 | D14s67 | (57) |
| IDDM12 | 2q33* | CTLA-4, CD28 | (58) |
| IDDM13 | 2q34 | D2s137, D2s164, IGFBP2, IGFBP5 | (59) |
| IDDM14 | ? | NCBI# 3413 | |
| IDDM15 | 6q21 | D6s283, D6s434, D6s1580 | (52) |
| IDDM16 | ? | NCBI# 3415 | |
| IDDM17 | 10q25 | D10s1750- D10s1773 | (60) |
| 2p12 | EIF2AK3 | | (61) |
| 5p11-q13 | | | (62) |
| 16p | | D16s405- D16s207 | (62) |
| 16q22-q24 | | D16s515- D16s520 | (55) |
| 1q42 | | D1s1617 | (63) |
| Xp11 | | DXS1068 | (64) |
In summary, T1DM is a complex, multifactorial disease involving genetic predisposition and an environmental triggering event, of which viral causes have been proposed. Although more than 50 loci have been identified, genes involved in immune regulation including HLA subtypes, VNTR in insulin itself, CTLA4, PTPN22, AIRE, and IL2R remain most prominent (65,66). The HLA association, especially class II, remains the strongest predictor of T1DM risk. The heterozygous DR3/DR4 genotype carries the highest genetic risk for T1DM in non-Hispanic whites (45-70). In conclusion, insulin expressing islets from recent-onset T1D subjects show overexpression of interferon stimulated genes (ISGs), with an expression pattern similar to that seen in islets infected with virus or exposed to IFN-γ/interleukin-1β or IFN-α.
Autoantigens and Autoantibodies in Type 1 Diabetes
The Doniach group in London, first reported islet cell autoantibodies in patients with autoimmune polyglandular syndromes (APSs) (71), especially in those with APS type-1 (APS-1) (72), even though such patients did not often develop diabetes. Lendrum and colleagues, having failed to find serological evidence for an autoimmune basis for chronic pancreatitis, did succeed in finding Islet Cell Antibodies (ICA) detectable by indirect immunofluorescence in patients with T1DM. Islet cell surface reactive autoantibodies and autoreactive peripheral blood T cells were also reported (73,74). Over the years that followed, the presence of ICA in US patients was confirmed but with distinctly lower frequencies of ICA among African American diabetic patients (75). Insulin autoantibodies (IAA) were discovered in patients with T1DM before their first dose of insulin replacement had been received (76). The presence of IAA together with ICA identified a group of non-diabetic relatives of probands with T1DM, that were at high risk for T1DM themselves (77). Insulin itself is not an ICA antigen that can be detected by the indirect immunofluorescent technique. Subsequently, much of the antigenic nature of the ICA reactivity has become clearer. It was recognized that many patients with "stiff" man syndrome who were prone to develop diabetes, also had ICA and autoantibodies to glutamic acid decarboxylase (GAD65). These GAD autoantibodies penetrated the blood brain barrier. High concentrations of GAD in the cerebellum reduce brain levels of the inhibitory neurotransmitter gamma aminobutyric acid (GABA), thereby causing the appearance of temporal lobe epilepsy, depressed cognition, muscle spasms, cerebellar incoordination and motor dysfunctions. That GAD65 was the antigen that accounted for the 64 KDa islet cell protein previously discovered by Baekkeskov to react with autoantibodies in T1DM, was later confirmed by the same investigator (78). Antibodies to recombinant GAD65 and GAD67 in T1DM patients were soon reported (79). The autoantibodies reacted to the antigens by conformational rather than linear epitopes, and thus with native rather than denatured antigens. Therefore, they were best detected by liquid phase assays such as radioimmunoassay, rather than by an ELISA technique. In stiff-man syndrome, the predominant GAD autoantibodies reacted with linear epitopes. It became known that besides islet cell 64 KDa sized proteins, autoantibodies in the sera of T1DM patients also precipitated islet cell proteins of 50, 40 and 37 KDa as well (80).
The next islet cell antigen discovered was one of the two-dozen tyrosine phosphatases expressed in islet cells, insulinoma antigen-2 (IA-2) (81). This antigen shared structural homologies with the ICA-512 antigen (82). A second tyrosine phosphatase named IA-2ß was discovered next (83). These additional tyrosine phosphatase antigens allowed for the matching of the islet cell proteins previously identifiable only by their molecular weights. Thus, GAD65 and its tryptic fragment explained the 64 and 50 KDa proteins, while tryptic fragments of IA-2 and IA- 2ß were identical with the 40 KDa and the 37 KDa islet precipitable proteins respectively (84). The tyrosine phosphatases are a family of transmembrane enzymes of which only these two are expressed by the pancreatic islets and react with T1DM autoantibodies. The reactivity is almost exclusively with the internal domains of these molecules, suggesting that they arise as a consequence of islet cell damage from autoimmunity. Antibodies to IA-2 cross-react with those of IA-2ß in about 50% of the patient sera. Some unusual patient sera however react exclusively with IA-2ß. The question of why only these two members of the tyrosine phosphatase family are targets of islet cell autoimmunity has been answered by the finding that they are relatively resistant to proteolytic enzymatic digestion, and once released from islet cells after their lysis, are insoluble and thus become better antigens for auto-immunization, than those that remain soluble and are more rapidly digested (85).
Recently, another antigen of 38KDa size (GLIMA) was added to the islet cell group, albeit only a minority of patient's sera reacts to it (86). Still more islet cell autoantigens are likely to be discovered. The detection of islet cell autoantibodies is useful for differentiating T1DM from diabetes of other causes, and can be used to predict onset of diabetes months to years before onset of the clinical disease (20,21,87,88) in non-diabetic relatives of probands with T1DM. Importantly, the clinical onset of the disease is often long preceded by the appearance of autoantibodies reactive to islet cells (ICA) (88) and to insulin (77), as independent age-related variables in predicting a diabetic outcome (89). Islet cell autoantibodies (ICA) also show a strong tendency to disappear after diabetes onset when all ß cells are destroyed (90,91).
Studies in mice demonstrated a critical role of autoantibodies to GAD65 in the induction of autoimmune diabetes in NOD mice. In humans, the German BABY-DIAB study and the Finnish TRIGR study showed that islet autoantibodies which are mostly IgG class can be transferred through the placenta from islet antibody-positive mothers to their offspring (92,93). Most of the antibodies, however, disappeared from the circulation of the infant within the first year of life, indicating that they represent maternal antibodies and unlikely that they are markers of fetal induction of B-cell autoimmunity (93). In the German BABY-DIAB study, it was demonstrated that 729 offspring of mothers with T1DM had significantly lower risk of developing multiple islet autoantibodies (5 year risk 1.3%) and diabetes (8-year risk 1.1%) when they were GAD or IA-2 positive, than offspring who were islet autoantibody negative at birth (94). These findings suggest that fetal exposure to islet autoantibodies may protect from future diabetes. Furthermore, the German BABY-DIAB study finding is consistent with the overall decreased risk of development of diabetes in offspring of mother with T1DM compared with that of offspring of fathers with T1DM and nondiabetic mothers (95).
The timing of the appearance of the autoantibodies seems to be important. It was found that progression to multiple islet autoantibodies was fastest in children who were antibody positive by age 2 years and that progression to diabetes was inversely related to the age of first positivity for multiple autoantibodies (96).
The presence of multiple autoantibodies strikingly increases the risk of diabetes, whereas one of the above autoantibodies in the absence of all of the others when tested for, denotes only a modestly increased risk (20,21). This suggests that antigenic epitope spreading is involved in a sustained or accelerated autoimmune attack (72) (97). Besides autoimmunity to islet cell autoantigens, patients with T1DM are subject to other autoimmunities. Thus T1DM is a component part of the autoimmune polyglandular syndromes, commonly in APS-2 (Diabetes Mellitus, Addison Disease, Hypothyroidism) and with less frequency in APS-1(AIRE gene mutations) (72). Accordingly, patients with T1DM have high rates of thyroid autoimmunity, especially if they are females (98) (99), and are at increased risk for Addison's disease (99), atrophic gastritis (100), pernicious anemia (98), celiac disease (101), and vitiligo (102).
Table 5.
Autoantibody Targets in Type 1 Diabetes
View in own window
| glutamic acid decarboxylase 65 |
| Islet cells |
| Insulin |
| Zinc Transporter 8 |
Antigen Specific Cellular Immunity in Type1 Diabetes
Autoreactive T cells that develop in impending T1DM, localize to the pancreatic islets where they become a component part of the evolving insulitis lesions. Thus, circulating autoreactive T cells are relatively sparse in impending T1DM. Nevertheless, antigen specific T cells are identifiable through prolonged in-vitro cultures in the presence of purified or recombinant islet cell autoantigens such as GAD (103) (104) and IA-2 (105). In fact, autoreactivity to a large number of autoantigens have been reported in both human and murine diabetes (106). T cell proliferative responses to insulin and GAD65, and more generally to islet extracts, have been repeatedly reported in both patients with T1DM (107,108) and NOD mice. However, both in humans and NOD mice, reports of spontaneous proliferative responses have been difficult to reproduce and validate, probably because of the relative paucity of autoreactive T cells in peripheral blood samples, and the ready contamination of recombinant "test" antigens by lymphotoxin or lipopolysaccharide (LPS), that by itself, can produce proliferative responses even when present in trace amounts. Furthermore, significant T cell responses to insulin, proinsulin or GAD65 antigen were reported, in some normal controls as well as in T1DM patients (109-111). Numerous laboratories have reported T cell reactivity in diabetic patients against GAD65 and IA-2 and their peptides with variable results (105,107,112-117). However, in established diabetes, the loss of the majority of ß cell mass resulting in associated loss of GAD65 and other ß cell antigens, in turn leads to the inactivation of T cells due to the loss of the peptide antigens that were driving the response. Thus, antigenic/epitopic spreading is an undesirable phenomenon associated with progression in autoimmune diseases like T1DM to a clinically significant outcome.
Pathogenesis of Type 1 Diabetes
The availability of Biobreeding (BB) rats and nonobese diabetic (NOD) mice, the rodent models of T1DM, has greatly enhanced our understanding of the possible pathogenic mechanisms involved (). Recently, it has become possible to compare these findings with findings in human islets, obtained from post mortem specimens of the pancreas through the network of Pancreatic Organ Donors (nPOD) and from patients with recent onset DM via endoscopic pancreatic biopsy (DiViD study, Norway) (86,118,119). In addition, epidemiological studies aimed at the prediction and prevention of T1DM permit a picture of the natural history to emerge. The process of destruction of β-cells is chronic in nature, often beginning during infancy and continuing over the many months or years that follow. At the time of clinical diagnosis of T1DM, about +80% of the β- cells have been destroyed, the islets are infiltrated with chronic inflammatory mononuclear cells (insulitis), including CD8+ cytotoxic T cells. Once islet cell autoimmunity has begun, progression to islet cell destruction is quite variable, with some patients rapidly progressing to clinical diabetes, while others remain in a non-progressive state.
Diabetes risk and time to diabetes in relatives of patients directly correlates with the number of different autoantibodies present. The pathogenesis of T1DM has been extensively studied, but the exact mechanism involved in the initiation and progression of β-cell destruction is still unclear. The presentation of beta cell-specific autoantigens by antigen- presenting cells (APC) [macrophages or dendritic cells (DC)] to CD4+ helper T cells in association with MHC class II molecules is considered to be the first step in the initiation of the disease process. Macrophages secrete interleukin (IL)-12, stimulating CD4 + T cells to secrete interferon (IFN)-γ and IL-2. IFN-γ stimulates other resting macrophages to release other cytokines such as IL-1β, tumor necrosis factor (TNF-α) and free radicals, which are toxic to pancreatic β-cells. During this process, cytokines induce the migration of β-cell autoantigen specific CD8+ cytotoxic T cells. On recognizing specific autoantigen on ß cells in association with class I molecules, these CD8+ cytotoxic T cells cause ß cell damage by releasing perforin and granzyme and by Fas-mediated apoptosis of the beta cells. Continued destruction of beta cells eventually results in the clinical onset of diabetes.
Recently, these concepts derived from studies in the rodent models have been challenged as having the same pathologic process that occur in humans. Analysis of variations in histopathology observed from these organ donors provide mechanistic differences related to etiological agents and serve an important function in terms of identifying the heterogeneity of T1D (120). The findings are not always consistent with those of the rodent models. For example, the dense infiltration of islets by T-cells is evident in the pancreas of those who succumb to DKA at onset, but more chronic cases show a patchy distribution of destroyed and functioning islets containing beta cells with insulin suggesting a defect in secretion rather than synthesis. In the DiViD (Diabetes Virus Detection) study, expression of inflammatory markers, predominance of Class I antigens (rather than expression of Class 2 antigens) in islets, and actual viral isolations suggest a more acute process. Taken together, the studies suggest that T1DM may be a heterogeneous group of conditions in which auto-immunity may be a consequence or companion rather than the initiating mechanism. These findings begin to explain why prediction of developing T1DM in those from affected families considered at risk has become quite accurate, whereas prevention or reversal of DM by immune intervention or modulation has failed repeatedly (3,4,121).
The Inductive Event in Type 1 Diabetes
Various mechanisms have been proposed:
MOLECULAR MIMCRY
In antigenic molecular mimicry, cross-reactive immune responses occur due to significant structural homologies shared by molecules encoded by dissimilar genes.
The incidence of T1DM has increased over the last three to four decades in Europe, and the clinical disease exhibits preferential seasonal onset (122). These observations emphasize the role of environmental factors in the disease process. It has long been suggested that T1DM in humans is caused by viral infections (123-125). However, despite a vast increase in the information regarding the various genetic factors controlling the disease, little is known about the role of the putative environmental factors that might provide a more direct approach to therapy (8). Specifically, allegations that childhood vaccines could be causal have not been upheld by more extensive controlled studies.
The disease pathogenesis may involve multiple factors including the genetics of the host, strain of the virus, activation status of the autoreactive T cells, upregulation of pancreatic MHC class I antigens, molecular mimicry between viral and ß cell epitopes and direct islet cell destruction by viral cytolysis. Viruses, as one of the environmental factors affecting the induction of T1DM, may act as triggering agents of autoimmunity or as primary injurious agents, which directly damage pancreatic ß cells. Immune responses against a determinant shared by host cells and a virus could cause a tissue-specific immune response by generation of cytotoxic cross-reactive effector lymphocytes or antibodies that recognize self-proteins located on the target cells.
Monoclonal antibodies against viruses have been observed to be capable of cross-reacting with host determinants (126).
Several studies in humans also point to viruses as triggers of the disease (127). Coxsackie B4 virus and rubella virus have been linked with T1DM. In a few instances, Coxsackie B4 virus has even been directly isolated from pancreatic tissues of individuals with acute T1DM. Inoculation of this virus into mice, in one report, produced diabetes (128). The possibility that viruses might cause some cases of T1DM by infecting and destroying pancreatic ß-cells has received considerable attention. However, it is difficult to demonstrate in-vivo that viruses replicate in human ß-cells and/or produce diabetes in man. An in-vitro system was therefore developed to determine whether viruses are capable of destroying human β-cells in culture (129,130). By this method, it was clearly shown that several common human viruses, including mumps virus (131), Coxsackie B3 virus(132), Coxsackie B4 virus (128), reovirus type 3 (133), could infect human ß-cells. In addition, by radioimmunoassay, it was shown that the infection markedly decreased the insulin content of the ß-cells.
A strong correlation was found between the CMV genome in the immunocytes and the islet cell autoantibodies in the sera from diabetic patients (134). About 15% of newly diagnosed autoimmune T1DM patients have been reported to have persistent CMV infections.
Furthermore, it has been proposed that a molecular mimicry between protein 2C (p2C) of Coxsackie virus B4 and the autoantigen GAD65 may play a role in pathogenesis of T1DM. Kaufman et al (135) and Vreugdenhil et al (125), showed that the amino acid sequence of p2C shares a striking homology with a sequence in GAD65 (PEVKEK) and is highly conserved in Coxsackie virus B4 isolates as well as in different viruses of the subgroup of Coxsackie B-like viruses. These are the most prevalent enteroviruses and therefore the exposure to the mimicry motif should be a frequent event throughout the life. Furthermore, they suggested that molecular mimicry might be limited to the HLA-DR3 subpopulation of the T1D patients.
Although numerous sequence similarities between viral proteins and ß-cell autoantigens are plausible, the relationship between Coxsackie virus infection and GAD65 autoimmunity has received the most attention.
Glutamate Decarboxylase (GAD)
The finding by Kauffman et al (135), of a striking sequence homology of 18 amino acid peptide between human GAD65 and the Coxsackie virus p2-C protein, enhanced the evidence of a specific molecular mimicry model involving GAD. In addition, this specific region of GAD65 contains a T cell epitope involved in the GAD cellular autoimmunity in humans with immune mediated diseases (103) and this region is an early target of the cellular immunity in NOD mice (136,137). GAD catalyzes the formation of the inhibitory neurotransmitter γ-amino butyric acid (GABA) from glutamine (104). Two forms of GAD exist (GAD65 and GAD67). GAD65 is the predominant form within the human pancreatic islet cells, while GAD67 predominates in mouse islets. Within the islets, GAD is predominantly observed within the ß-cells, while its roles in the inhibition of somatostatin and glucagon secretion and in the regulation of proinsulin synthesis and insulin secretion, have also been suggested (138).
Another study further supports a link between Coxsackie virus and T1DM, associating IgM antibodies to Coxsackie B virus as a marker of recent exposure to the virus in newly diagnosed IMD patients and age/sex-matched controls (139). In that report, humoral immunity to Coxsackie virus and GAD appeared to cluster, even in people without diabetes. A series of overlapping synthetic GAD65 peptides were used to study the most reactive T cell determinants in individuals at increased risk for T1DM, i.e., autoantibody positive, first degree relatives of T1DM patients. Elevated in vitro T cell responses were observed to GAD65 peptides (amino acids 247-266 and 260-279) in newly diagnosed T1DM patients and autoantibody positive at- risk individuals (140). The sequence of this region of GAD65 (amino acids 250-273) is significantly similar to the p2-C protein of Coxsackie B virus (123). However, not all published reports have demonstrated a linkage between immunity to GAD and Coxsackie virus. For example, one study identified a non-Coxsackie-homologous region of GAD65 as a predominant cellular immune epitope while studying the polyclonal human T cell responses (115).
Insulinoma Antigen Two (IA-2)
Tyrosine phosphatase IA-2 is another molecular target of pancreatic islet autoimmunity in T1DM. In one recent study, the epitope spanning 805-820 amino acid elicited maximum T-cell responses in all at-risk relatives, out of a total of 68 overlapping, synthetic peptides encompassing the intracytoplasmic domain of IA-2 (141). This epitope was found to have 56% identity and 100% similarity over 9 amino acids with a sequence in VP7, a major immunogenic protein of human rotavirus. This dominant epitope also has 75-45% identity and 88-64% similarity over 8-14 amino acids to sequences in Dengue, cytomegalovirus, measles, hepatitis C and canine distemper viruses and the bacterium Haemophilus influenzae.
Furthermore, three other IA-2 epitope peptides have 71-100% similarity over 7-12 amino acid stretch to herpes, rhino-, hanta- and flavi-viruses. Two others have 80-82% similarity with dietary proteins of milk, wheat and bean proteins. These molecular mimicries could lead to triggering or exacerbation of ß-cell autoimmunity.
SUPERANTIGENS
Besides molecular mimicry, retroviral expression of superantigens (Sags) may be able to activate clonal expansion of autoreactive T cell clones. Superantigens have been implicated in the pathogenesis of the various autoimmune diseases (142,143). Originally described as minor-lymphocyte stimulating antigens, retroviral Sags expressed by B cells interact with the development of T helper cells of both Th1 and Th2 subtypes in mice. A study in patients with T1DM demonstrated that two thirds of IAA positive sera also reacted with p73 (144). Conrad et al (145) isolated a novel mouse mammary tumor virus-related human endogenous retrovirus (HERV), in patients suffering from acute onset T1DM. He termed them the HERV IDDMK1,2 22 subtype. They further showed that the N-terminal moiety of the envelope (env) gene encoded an MHC class II-dependent superantigen. He proposed that expression of this Sag, induced extra-pancreatically and by professional antigen-presenting cells, could lead to ß-cell destruction via the systemic activation of autoreactive T cells. He further reported the selective expansion of Vß7+ T cells in the islet cell infiltrates from two patients with recent onset IMD was associated with extensive junctional diversity of Vß7+ T cell clones. These investigators demonstrated that islet cell membrane preparations preferentially expanded Vß7+ T cells from non-diabetic peripheral blood mononuclear cells (146). However, other investigators were unable to confirm T1DM specificity of the IDDMK1,2 22, since it was equally recoverable as viremia from controls as well as patients (147). Furthermore, both patients and controls made antibodies to env proteins.
In order to establish molecular mimicry as a mechanism responsible for the autoimmune diseases it is important to identify the precise epitope that initiates the putative cross-reactive immune response. Additional complexity that has come to various animal studies is that of epitope spreading (148). An increasing array of autoantigens or autoantigenic peptides reactive with autoantibodies develop over time. Both intramolecular and intermolecular epitope spreading has been described in NOD mice (136,149). These studies demonstrated that T- cell responses in NOD mice expand in vivo against a defined group of islet cell antigens in an orderly sequential manner. These responses in the young NOD mice first show a strong reactivity to GAD enzyme and not to other islet cell antigens. Furthermore, the initial response to GAD is first limited to one region of the protein only. Gradually, this response spreads intramolecularly to involve other regions of the protein. Eventually, after the destructive islet cell inflammation (insulitis) as a result of autoimmunity to ß-cells, the T-cell responses spread intermolecularly to involve other islet cell proteins (e.g., heat shock protein 60, carboxypeptidase H and insulin) as well (150). This epitope spreading makes it difficult to predict which putative cross-reactions, if any, are important in terms of disease induction, and which do not give rise to autoimmune pathology, particularly in humans who are exposed to many infections.
Deficiencies in immunoregulation in Type 1 Diabetes
There is both evidence for and speculation about defective central and peripheral mechanisms of immunoregulation in the autoimmune form of T1DM. Deletion of autoreactive T cells in the thymus, is one mechanism for the induction of tolerance to self-antigens (central deletion). This may involve diminished expression of insulin in the thymus of susceptible individuals due to the presence of class I VNTR alleles 5' to the insulin gene as already discussed. Others have suggested that it is the ineffective antigenic binding of the T1DM-prone HLA-DQ or -DR that promotes islet cell autoimmunity, since this permits autoreactive T cells to escape thymic ablation and pass into circulation.
In addition to clonal T cell deletion and anergy in thymus, peripheral regulatory T (Treg) cells are essential for the down regulation of T cell responses to both foreign and self-antigens, and for the prevention of autoimmunity. Various studies have identified defects in the peripheral Treg cells in T1DM patients (151,152) as well as in NOD mice affecting both NKT cells (153,154) as well as CD4+CD25+ suppressor T cells (155). Since these Treg cells are not absent in either species, ways to stimulate them should be actively sought to provide novel therapies for the future. The possibility of future therapeutic use of Treg cells in human autoimmune diseases lies heavily on basic studies that are designed to elucidate the mechanisms of induction and function of these cells. Therapy with immunomodulatory compounds that specifically target endogenous pools of Treg cells can be envisioned (156). This approach requires a more detailed investigation into the intracellular and extracellular events that regulate the differentiation and expansion of these cells in-vivo.
Of great interest has been the emergence of immune mediated T1DM in patients treated with checkpoint inhibitors for various cancers (157). Unlocking the immune response via drugs that block the molecules programmed death (PD1) or its ligand, PDL1, as well as CTL4, may result in immunotoxicity with emergence of autoimmunity affecting various organs, including endocrine tissues such as the thyroid, adrenal and pancreas causing a form of T1DM (158). Indeed, autoimmunity has been called the “Achilles’ Heel” of immunotherapy, with increasing reports of its association with T1DM (159).
Environmental Factors in Type 1 Diabetes
Besides the familial predispositions, much evidence points to a major role of environmental factors in the disease pathogenesis. More than 60% of identical twins affected by T1DM are discordant for the disease and most of the non-diabetic twins lack islet cell autoantibodies. Over the past 3 decades, the disease frequency is on a steep rise in Western countries that cannot be explained by the accumulation of the susceptible genes. Africans, who dominate the tropics, and Chinese, both have low frequencies of the susceptible genes and low incidence rates of T1DM (75), except where there has been a high rate of Caucasian genetic admixture.
More persuasively, migrants from countries with low hygiene and low incidence rates of T1DM to countries with high hygiene and high incidence become as susceptible as the natives within a generation (160). Animals reared in sterile environments have early onsets and increased frequencies of diabetes while those infected with a variety of micro-organisms and parasites become protected (161-165). The hygiene hypothesis was proposed. A strong causal relationship between prevailing level of community hygiene, especially with respect to drinking water and the dramatic increase in the incidence of autoimmune diseases such as T1DM in the modern world, has been referred to as the hygiene hypothesis.
ROLE OF DIET
Despite persuasive epidemiological evidence for environmental factors that precipitate T1DM in genetically susceptible individuals, their identity remains elusive. This may be due to long period between exposure and the onset of hyperglycemia, the complex genetics of the disease, and the likely multiple insults of perhaps different derivation involved in the initiation of the insulitis and subsequent ß cell destruction. Dietary habits such as consumption of dairy products and early weaning of infants, and dietary toxins such as nitrates and nitrites have been associated with this autoimmune disease (166,167).
Close correlations between per capita consumption of unfermented milk proteins and the incidence of diabetes between countries(168-170) and within a country have been reported (171). The claimed negative association between diabetes incidence and a high frequency and long duration of breast-feeding is more controversial (166) and has not been confirmed by reports from Germany (172) and the United States. Several studies have found associations between the consumption of foods rich in nitrates (or nitrites), which is reduced to nitrite in the gut, and the occurrence of T1DM (173,174). The active species is believed to be N-Nitroso compounds that can be formed from the reaction of nitrite with amines (175). Most recently, the gut microbiome and its modulation by dietary factors, has been implicated in the causality of T1DM (176).
The incidence of T1DM varies worldwide according to dietary patterns. In-depth exploration of dietary risk factors during pregnancy and early neonatal life is warranted to confirm whether and to what extent diet cooperates with genetic susceptibility in the early onset of T1DM.
Screening Methods for Type 1 Diabetes
T1DM is by far the most common chronic metabolic disease of childhood and adolescence and its prevalence and incidence has been increasing worldwide (96). This increase of incidence is the highest among the children under 5 years of age (177). Prevention of T1DM would constitute a major advance in the lives of pre-diabetic individuals and significantly relieve a major current and predicted burden on both the individual and the health care system. Identifying individuals at risk developing the disease and the prevention of the disease progression are two important steps before the onset of disease. The presence of islet autoantibodies, as well as the genetic predisposition with specific HLA haplotypes are known risk factors associated with the development of diabetes. Most studies have been carried out on first-degree relatives of T1DM patients who have 15-fold increased risk of the developing diabetes in comparison to the general population. However, more than 90% of all patients developing T1DM do not have an affected family member. Therefore, it is crucial to establish a standardized screening method which will efficiently identify individuals at high risk in a general population. School children between 5-18 years of age were screened to evaluate the predictive value of autoantibodies over a period of 6-12 years (178). This study indicated that the risk of developing T1DM when ICA is detected in the absence of other autoantibodies is low, whereas with more than one autoantibody (either GAD65A, IAA, IA-2A or IA-2ßA) the risk of developing T1DM in a general population is high. Similar findings were also reported in other studies (179-181). These results support the value of multiple autoantibodies as good predictive markers for T1DM not only in first degree relatives but also in the general population. Consequently, the American Diabetes Association now considers the presence of 2 or more autoantibodies as form of early presymptomatic diabetes (182).
Prevention Trials in Type 1 Diabetes
The elucidation of the natural history of pre-diabetes has allowed for the characterization of those individuals at greatest risk for developing autoimmune T1DM, through the use of genetic, immunologic and metabolic markers. This predictive ability has become possible in both high- risk relatives and the general population as mentioned above. The subclinical autoimmune destruction of ß-cells in the pancreas may last from a few months to several years. This pre- diabetic period has allowed investigators to test prevention strategies, which mainly have focused in modulation of autoimmune process (183). A number of studies initiated with general immunosuppressive agents, such as cyclosporin-A, azathioprine and prednisone in patients with new clinical onset T1DM, positive results in that insulin free remission rates were increased and endogenous insulin (C-peptide) reserves were improved (121). However, despite continued immunotherapy with the attendant risks of renal damage and lymphomas at higher doses, relapses proved to be the rule and such treatments were abandoned. Cyclosporin given at a prediabetic phase of the disease delayed but did not prevent diabetes (184,185).
With the observation that nicotinamide prevents pancreatic ß cell destruction from streptozotocin by raising otherwise depleted levels of islet cell NAD as a result of superoxide induced DNA breaks and repair, the vitamin was subjected to a large European and Canadian trial called The European Nicotinamide Diabetes Intervention Trial (ENDIT). However, nicotinamide failed to prevent progression to diabetes (186). In addition, a study in Germany (DENIS) was completed without any effect of nicotinamide on prevention of T1DM.(187).More recent studies have used Anti CD21(Rituximab), Anti CD3, Anti CTLA-4, oral insulin,GAD65 peptides, and infusions of Treg cells with early encouraging results in preserving insulin secretion, but without durable effects (188). These results in humans were often based on animal studies in NOD mice (189-191). In stark contrast to these encouraging studies in NOD mice, where a variety of interventions induce long lasting remissions, none of the studies in humans has so far yielded long-lasting remissions in humans (183,188).
Table 6.
View in own window
| Study and Phase | Drug | Age | Eligibility | Ref |
|---|
| TRIGR | Cow’s milk hydrolysate | 0-7 days | First Degree relatives, High-risk HLA | (192) |
| BABY DIET | Gluten-free diet | Younger than 3 months | Relatives, high risk HLA DR, DQ | (193) |
| TrialNet NIP | Docosahexaenoic acid | >24 weeks gestation- newborn | Relatives, HLA DR3 or DR4 | (194) |
| TrialNet Teplizumab | Teplizumab | 8-45 years | At least 2 confirmed autoantibodies and abnormal glucose tolerance | (195,196) |
| DIAPREV-IT | GAD-alum | 4-18 years | Islet autoantibody positive | (197) |
| TrialNet Oral Insulin, Phase III | Human insulin | 1-45 years | Relatives, 2+islet antibodies including to insulin | (198) |
| INIT I/II, | Intranasal insulin | 4-30 years | Relatives, 2+islet antibodies, HLA not DR2, DQ6 | (199) |
| Pre-Point, Phase I/II | Human insulin | 1.5-7 years | First degree relatives,
>50% risk of T1DM | (200) |
| FINDIA | Insulin-free whey- based formula | Infants | General population, high-risk HLA DQ | (201) |
| Teplizumab | Teplizumab | </=18 years of age | Relatives | (202) |
| Golimumab | Golimumab | 6 to 21 years | Newly diagnosed T1DM | (203) |
ATYPICAL DIABETES
Genetic Defects of ß-cell Function (Monogenic Diabetes)
Monogenic forms of diabetes are characterized by impaired secretion of insulin from pancreatic β cells caused by a single gene mutation. These forms comprise a genetically heterogenous group of diabetes including, maturity onset diabetes of the young (MODY), permanent or transient neonatal diabetes (NDM), and mitochondrial diabetes. MODY is the most common form of monogenic diabetes, with autosomal dominant transmission of a gene encoding a primary defect in insulin secretion (232-235).
Approximately 1 to 2% of diabetes in Europe is MODY (236). The clinical characteristics of these patients are heterogeneous, and not reliable in predicting the underlying pathogenesis (237,238). It is often misdiagnosed as T1DM or T2DM. Several genetic abnormalities have been found that account for the disorder. Some members of an affected family may have the genetic defect but not develop the diabetes phenotype. Whether this is due to modifying genes or environmental factors is unclear. MODY differs from the classical immunological T1DM in several ways. With MODY, a dominant family history of diabetes (if known) is always present. However, de novo mutations can occur. Hyperglycemia is mostly mild with a minimal tendency to ketosis before the age of 25 years, the insulin secretion in response to oral (OGTT) or intravenous (IVGTT) glucose administration is modestly decreased, and evidence of islet cell autoimmunity is absent. It is estimated that more than 80% of patients with monogenic diabetes are either not diagnosed or are misclassified as type 1 or type 2 DM (239).
The underlying genetic defects of the many MODY subtypes have been identified, as indicated below (Table 11). To date, fourteen genetic forms of MODY are recognized. MODY resulting from defects in the glucokinase gene (GCK) and hepatocyte nuclear factor-1-alpha (HNF-1α) are the most common types seen during childhood (MODY-2) and post puberty (MODY-3), respectively. MODY Types 2 and 3 together constitute 80% of all cases of MODY syndromes.
MODY 2 is the most common form of MODY with a prevalence of about 1:1000 people. It is caused by a dominant heterozygous inactivating mutation in glucokinase, the enzyme that phosphorylates glucose to permit its oxidation to ATP and hence insulin release. Insulin is released but at higher glucose concentration-the curve is right shifted but otherwise normal. Thus, fasting glucose is in the range of ~95-110 mg/dl and may remain above 140 mg/dl at 2 hours post prandial but returns to normal thereafter. HbA1c is in the range of 5.8-7.6% and generally remains in the low- mid 6% range. Patients are rarely symptomatic and may be discovered by chance when a blood glucose is obtained. Treatment is not necessary except during pregnancy in some cases; there is a very low prevalence of micro-macrovascular disease even after almost 50 years of follow-up. Young women are often discovered to have mid hyperglycemia when tested during pregnancy and erroneously labeled as having gestational diabetes. The non-affected fetus of an affected Mother may have some macrosomia in utero-the result of extra insulin secretion by the fetus in response to the maternal hyperglycemia (240).
MODY3 is the next most common form of MODY caused by a heterozygous mutation in HNF-1α, necessary for normal insulin secretion. Onset is usually in the teen years and glucose is in the mid-200s with mild to moderate symptoms. Patients may respond to sulfonylurea drugs initially, but later may go on to insulin dependence and more severe hyperglycemia. As with other MODY forms, a family history of diabetes is often obtained, with a diagnosis of T2DM common for older patients and T1DM in younger patients. Confirmation of the diagnosis by molecular testing is essential for recommending treatment and family counseling (241).
Defects in four pancreatic ß cell-specific transcription factor genes, HNF-1β (MODY5), HNF-4α (MODY1), pancreatic and duodenal homeobox 1 gene (PDX1) [previously termed insulin promoter factor-1 (IPF-1)] (MODY4) and neurogenic differentiation 1 gene (NeuroD1) and BETA2 (MODY6) are responsible for others. In contrast to MODY-2, patients with heterozygous mutations in the HNF1A, HNF4A, or HNF1B and more rarely in PDX1 or NEUROD1 have progressive deterioration in glucose tolerance and are at risk for developing complications of diabetes (242).
More recently, mutations in the tumor suppressor protein KLF-11 (MODY7), the carboxyl ester lipase CEL (MODY8), the transcription factor, paired box gene 4, PAX-4 (MODY9), the insulin gene, INS (MODY10), and tyrosine kinase, B-lymphocyte specific gene, BLK (MODY11) have been described. MODY 12 and MODY 13 are due to mutations in the ABCC8 and KCNJ11 genes, respectively. Mutations in these 2 genes also have been reported in neonatal diabetes. They are very rare and represent fewer than 1% of all MODY cases.
Table 11.
View in own window
| MODY Type | Gene
Gene Loci | Incidence | Age at Diagnosis | Primary Defect | Associated Features | Severity of Diabetes | Ref |
|---|
| 1 | HNF-4α 20q | Rare | Postpubertal | Transcription gene defects in ß-cells lead to impaired metabolic signaling of insulin secretion. | - | Severe | (242) |
| 2 | Glucokinase
7p | 10-60% | Childhood | impairment of ß-cells sensitivity to glucose and; defect in hepatic glycogenesis | Reduced birth weight | Mild | (243) |
| 3 | HNF-1α
12q | 20-60% | Postpubertal | Similar to MODY1 | Renal glucosuria | Severe | (242-246) |
| 4 | PDX1 (IPF-1)13q | Rare | Early adulthood | Defects in transcription factors during embryogenesis lead to abnormal ß-cell development and function | - | Mild | (247) |
| 5 | HNF-1β 17cen- q21.3 | Unknown | Postpubertal | Similar to MODY 1 and 3 | Glomerulocystic kidney disease, female genital malformations, Hyperuricemia, abnormal liver function tests | Mild | (248) |
| 6 | NeuroD1/BETA2
2q32 | Rare | Early adulthood | Defect in this gene causes abnormal development of ß cell and function | - | Unknown | (249) |
| 7 | KLF11
2p25 | Very Rare | Early adulthood | Reduced glucose sensitivity of the beta cell | Phenotype similar to T2D | Unknown | (250) |
| 8 | CEL
9q34 | Very Rare | <20 years | Impaired endocrine and exocrine pancreatic function | Exocrine pancreatic dysfunction | Unknown | (251) |
| 9 | PAX4
7q32 | Very Rare | <20 years | Impaired gene transcription in pancreatic beta cells on apoptosis and proliferation | - | DKA is possible | (252,253) |
| 10 | INS
11p15.5 | Very Rare | <20 years | Defect in this gene may result the loss of beta cell mass through apoptosis | - | Unknown | (254) |
| 11 | BLK
8p23 | Very Rare | <20 years | decreases insulin synthesis and secretion in response to glucose by up- regulating transcription factors | Higher incidence in obese individuals | Unknown | (255) |
| 12 | ABCC8
11p15.1 | < 1% | <35 years | Inactivating mutations cause impaired secretion mild mode | | | (255) |
| 13 | KCNJ11
11p15.1 | <1% | | | | | |
| 14 | APPL1
3p14.3 | <1% | | adapter protein, phosphotyrosine interacting with pH domain and leucine zipper | | | (256,257) |
Neonatal Diabetes
Neonatal diabetes is a rare disorder with an incidence of 1:100,000-1:200,000 live births (232,258). It presents in first 6 months of life and its’ severity depends on the underlying mutation in that it is either transient or permanent. Almost 50% of cases with neonatal diabetes are permanent (PND) while the remainder are “transient” (TNDM) in that they remit, but may reappear and become apparent later in life or at times of stress. Heterozygous activating mutations in KCNJ11 and ABCC8 —which encode the Kir6.2 and SUR1 subunits, respectively, of the ATP-sensitive potassium channel, are the most common causes of PND. Missense mutations in the INS gene are also identified in patients with PND and they may have an autosomal dominant or recessive inheritance pattern (232,254,258). Genetic diagnosis is important since the KCNJ11 and ABCC8 mutations respond to treatment by sulfonylureas, possibly without need for additional insulin therapy because these drugs can close the β cell potassium channel by an ATP-independent route (259). It is increasingly apparent that the same mutations can become manifest for the first time well beyond infancy and diagnosed as T2DM or rarely T1DM. Severe mutations in the KATP genes, especially KCNJ11 also may present with a neurological component in a syndrome known as DEND (Developmental delay, Epilepsy, Neonatal Diabetes); early diagnosis and treatment with sulfonylurea drugs is reported to ameliorate the neurological manifestations as the KATP channels are expressed in the brain. The major form of transient neonatal diabetes results from anomalies of the imprinted region on chromosome 6q24,but mutations in KCNJ11 or ABCC8 can also cause TNDM (232). Various rare forms of syndromic disease which include NDM are described; early diagnosis may diminish or delay the hitherto described natural history and consequences (258).
Mitochondrial Diabetes
Point mutations in mitochondrial m.3243A→G cause another form of diabetes with an insulin secretory defect that is commonly associated with neuro-sensory hearing impairment and a strict maternal mode of inheritance (260). In addition, genetic abnormalities that result in the inability to convert pro-insulin to insulin (261), or the production of mutant insulin molecules (262), are other examples of specific genetic defects in ß cell function which are rare causes of diabetes.
Chronic Illnesses
Hemochromatosis is a progressively more common recognized cause of diabetes with aging, and does not present in a pediatric age group. However repeated blood transfusions for conditions such as thalassemia major can lead to diabetes associated with hemosiderosis.
Many patients with cystic fibrosis develop a form of T1DM often during their teenage years which may require insulin replacement and is labeled “cystic fibrosis related diabetes (CFRD)” (263). Most CF patients now live long enough for this to have become a more common problem with impact on overall well-being and severity of symptoms ascribed to CF and partially responsive to insulin therapy. DKA is rare in CFRD, perhaps because of the concurrent effects on the α-cell secreting glucagon as well as the β-cell secreting insulin. Patients with Gitelman’s syndrome develop diabetes which resolves when they are adequately replaced with magnesium, excessively lost through the kidneys in this syndrome. Gitelman syndrome is a recessively inherited genetic entity, but the presentation of DM is usually not until later midlife (264).
Genetic Defects in Insulin Action
There are a series of rare genetic abnormalities in the insulin receptor, or in the signal transduction events which follow insulin docking to its receptor resulting in diabetes. The recessive DNA breakage disease (Bloom’s syndrome) is associated with mild diabetes due to severe insulin resistance, with very high levels of circulating insulin and insulin like growth factor one (IGF-1). Progeria and lipodystrophy are other such causes (232). In the latter case, the absolute deficiency of leptin leads to uncontrolled lipolysis resulting in severe insulin resistance, which is partially reversible by leptin administration (232)/
Endocrinopathies Associated with Hyperglycemia
Several hormones, such as epinephrine, glucagon, cortisol, and growth hormone, antagonize the action of insulin. Whereas release of these hormones constitutes the protective counter regulatory response to hypoglycemia, primary over secretion of these hormones can result in glucose intolerance or overt diabetes.
Cushing's syndrome, due to pituitary and ACTH secreting adenomas or adrenal hyperplastic disease or to exogenous glucocorticoid administration, can lead to diabetes (
265). Steroid-induced diabetes is most often seen when there is pre- existing insulin resistance or a defect in insulin synthesis/secretion unmasked by the inability to increase insulin secretion to overcome the resistance to its actions induced by glucocorticoids.
Acromegaly is associated with overt diabetes in 10 to 15% of cases, and impaired glucose tolerance in a further 50% (
266,
267). In acromegaly, there is marked insulin resistance and hyperinsulinemic responses; DM occurs only when the hyperinsulinemic response cannot match the requirement to overcome the degree of resistance.
Pheochromocytomas are associated with both inhibition of insulin secretion and an increase in hepatic glucose output (
268). These changes lead to impaired glucose tolerance, the severity of which is directly related to the magnitude of catecholamine production (
269). When seen in children, these are usually a component of the Von Hippel-Lindau syndrome, MEN2, and NF1.
Glucagon-secreting tumors (glucagonoma) are associated with an unusual constellation of clinical features, including skin rash, weight loss, anemia, and thromboembolic problems. Approximately 80% of these patients have either impaired glucose tolerance or diabetes (
270).
Somatostatin-secreting tumors (somatostatinomas) are typically associated with the triad of diabetes mellitus, cholelithiasis, and diarrhea with steatorrhea (
271).
Although thyroxine is not a counter regulatory hormone, hyperthyroidism can interfere with glucose metabolism. It is associated with both increased sensitivity of pancreatic ß cells to glucose, resulting in increased insulin secretion, and antagonism to the peripheral action of insulin. The latter effect usually predominates, leading to impaired glucose tolerance in some untreated patients (
272).
Drug- or Chemical-induced Diabetes
A large number of drugs can impair glucose tolerance; they may act by decreasing insulin secretion, increasing hepatic glucose production, and/or by causing resistance to the action of insulin (273). Included in this list are several classes of antihypertensive drugs, such as beta blockers (274), protease inhibitors used for the treatment of HIV infection (275), and tacrolimus and cyclosporine used primarily to prevent transplant rejection (276,277). Drugs of the serotonin re-uptake inhibitor (SSRIs) class can lead to obesity, impaired glucose intolerance and T2DM, especially if individuals were already insulin resistant before they started such medications.
There is a common association between obesity, insulin resistance, hypertension, and dyslipidemia, which has been called syndrome X or the metabolic syndrome (207,212,278,279). The administration of a thiazide diuretic or a ß-blocker to such patients can exacerbate the insulin resistance and may bring on hyperglycemia (274). In comparison, angiotensin-converting enzyme (ACE) inhibitors and alpha-adrenergic antagonists (such as doxazosin) may improve insulin sensitivity. Because the former also protect against renal disease, they are the drugs of choice for diabetic patients with hypertension.
Viral Infections
Certain viruses e.g., Coxsackie B4, have been implicated to cause diabetes, either through direct ß cell destruction or possibly by inducing autoimmune damage. The direct proof of this however remains tenuous. Chronic hepatitis C virus infection is associated with an increased incidence of diabetes, but it remains uncertain as yet if there is a cause-and-effect relationship.