ABSTRACT
Mycobacterium tuberculosis, the etiological agent of tuberculosis (TB), is responsible for the largest number of deaths worldwide caused by a single organism. Over 25% of the world population is infected with M. tuberculosis, though active infections account only for a small percentage. Though some degree of endocrine dysfunction is invariable in all patients with TB, clinically significant endocrinopathy other than glucose intolerance is rare. This chapter reviews endocrine dysfunction and endocrinopathies associated with TB infection related to the adrenal, thyroid and pituitary glands. Additionally, functional derangement of sodium and calcium homeostasis is also covered. Adrenal involvement can be found in up to 6% of patients with active TB, however isolated adrenal involvement is seen only in a fourth of these. The most common clinical manifestation is Addison’s disease (AD). Clinical manifestations of AD appear only after 90% of the adrenal cortices have been compromised. Thyroid tuberculosis (TTB) is very rare, even in countries with a high prevalence of TB. TB has been seen to involve the thyroid in 0.1 to 1% of patients. Primary pituitary TB (in the absence of systemic involvement and/or constitutional symptoms) is extremely rare, and secondary pituitary TB is more commonly encountered in clinical practice. Pituitary TB should be considered in the differential of a suprasellar mass especially in developing countries, as the condition is potentially curable with treatment. Hyponatremia has been commonly seen in patients admitted to the hospital with TB. The commonest cause of hyponatremia is the syndrome of inappropriate antidiuresis (SIAD). Other causes include untreated primary or secondary adrenal insufficiency, volume depletion, hyponatremia associated with volume excess and hypoalbuminemia and rare cases of cerebral salt wasting seen with tuberculous meningitis. The prevalence of hypercalcemia in patients with TB has ranged from 2-51% in various studies. The primary determinant in the development of hypercalcemia among patients with TB appears to be their Vitamin D status and nutritional calcium intake. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Mycobacterium tuberculosis the etiological agent of tuberculosis (TB) was directly responsible for 1.3 million deaths in 2019. A majority of these deaths happen in patients without human immunodeficiency virus (HIV) co-infection making M. tuberculosis the pathogen responsible for the largest number of deaths in the world by a single organism. Additionally, TB is among the top ten causes of death worldwide (1).
Most cases of primary TB infections are clinically, bacteriologically, and radiologically inapparent. This primary infection in 5-10% patients leads to active disease after a period of latency within 2 years of contracting the infection. In another 5% the disease becomes active much later in life after a decline in general immunity. It is thought that over 25% of the world’s current population is infected with M. tuberculosis though active infections account only for a small percentage. In the year 2019 over 10 million patients were newly diagnosed with clinical TB. South East Asia accounted for over 44% of these along with Africa (25%), Western Pacific (18%), Eastern Mediterranean (8.2%), Americas (2.9%) and Europe (2.5%). The eight countries of India (26%), Indonesia (8.5%), China (8.4%), Philippines (6.0%), Pakistan (5.7%), Nigeria (4.4%), Bangladesh (3.6%) and South Africa (3.6%) account for two thirds of the world’s newly diagnosed cases last year (1).
As previously noted most active TB infections are reactivation of latent primary TB though a small but significant percentage of patients have active TB related to new exogenous re-infection. The most common primary site of adult active TB are the highly aerated upper lobes of the lungs. The defining pathology includes the presence of granulomas containing epithelioid cells, Langhan’s giant cells surrounded by lymphocytes with a center of caseous necrosis and varying degrees of fibrosis. This chapter focuses on the endocrinology of tuberculous infection (2, 3).
ALTERED IMMUNE-NEUROENDOCRINE COMMUNICATION IN TUBERCULOSIS
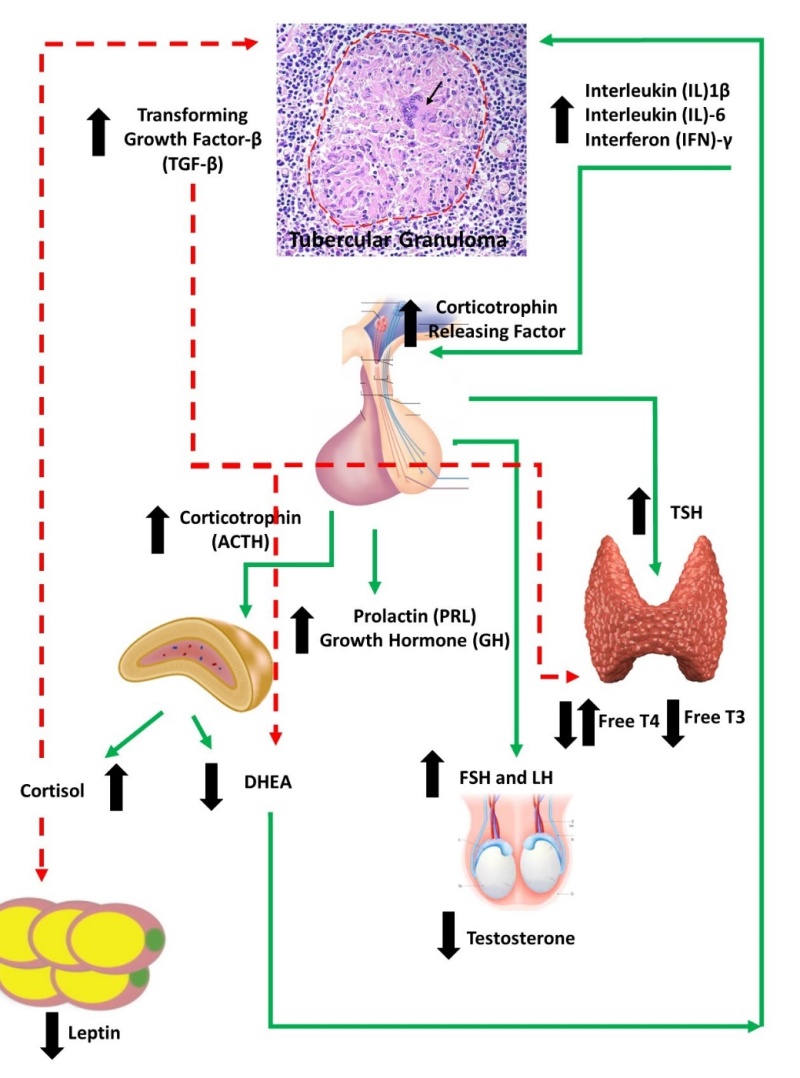
The two-way communication between the immune system and the neuroendocrine system is well known and documented. An activated immune cascade can affect all the endocrine systems of the body. Adrenal steroids are the primary hormones that modify immune responses. The up-regulation of the hypothalamic-pituitary adrenal (HPA) axis by inflammation related to infections is primarily mediated by the action of inflammatory cytokines on the hypothalamic releasing factors. Cytokines like Interleukin-6 (IL-6), Interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) stimulate the secretion of corticotrophin releasing hormone (CRH) from the hypothalamus leading to corticotrophin (ACTH) secretion from the pituitary. The action of ACTH on the adrenal cortex leads to secretion of both cortisol and dehydroepiandrosterone (DHEA). Cortisol inhibits the T- lymphocyte mediated Th1 response while DHEA antagonizes the cortisol action on Th1 response. This intense immune-endocrine response to acute infection leads to early mobilization of the immune cells and a robust immune response by the host against the offending pathogen (4, 5).
However, in TB the chronic persistent activation of the immune-endocrine axis leads to misuse of the immune-endocrine axis and can exacerbate damage to the host. Primarily the prolonged activation of the HPA and resultant increase in glucocorticoid (GC) secretion leads to a change in the T-lymphocyte response from Th1 to Th2 response (6). Beyond this GCs can interfere with gene expression of certain transcription factors like nuclear factor kappa-β (NF-κβ) (7), inhibit the proliferation of effector T cells and cause an increased rate of apoptosis of the regulatory T cells (8). Clinical studies in patients with TB have shown increased circulating levels of cytokines like IL-6, IL-10, interferon-α (IFN-α) and cortisol. However, DHEA levels have been consistently shown to be well below normal levels. In summary, GCs appear to have an adverse effect on the anti-TB immune response while DHEA appears to have a favorable effect. This balance is adversely impacted with the chronic inflammation seen in TB.
Some of these changes in immune-endocrinology have also been implicated in the morbidity associated with TB. In vitro studies suggest that negative immune response to mycobacterial antigens was associated with increased IL-6 production which in turn was associated with lower body weights among patients with TB. Higher circulating IL-6 was also associated with loss of appetite (9). The increased circulating GCs additionally mobilize peripheral lipid stores and inhibit protein synthesis and favor loss of lean body mass. Hypothalamic CRH secretion also appear to have direct catabolic effects on the body other than its effect mediated through increased GC secretion (10). Among the adipocyte hormones there is a decrease in leptin and increased secretion of adipocytokines in TB. In an acute infection the above adaptive response appears to be useful by directing limited energy stores to the immune response away from the body’s physiological needs. However, in chronic infections like TB these changes lead to a chronic metabolic deficit leading to cachexia which in turn then affects the further ongoing immune response and disease outcome (11, 12).
Some of these alterations in the immune-endocrine axis in M. tuberculosis infection are summarized in .
ENDOCRINOPATHIES IN PATIENTS WITH TUBERCULOSIS
Though some degree of endocrine dysfunction is invariable in all patients with TB, clinically significant endocrinopathies other than glucose intolerance is rare. In a small study of 50 patients hospitalized with sputum-positive pulmonary TB in South Africa the commonest endocrine dysfunction noted was a low free T3 state as part of sick euthyroid syndrome in over 90% of patients. The other common endocrine dysfunction noted in the study was a 72% prevalence of hypogonadotropic hypogonadism among male patients and a 64% prevalence of hyponatremia of whom almost half of them (17/50) had documented syndrome of inappropriate diuresis (SIAD). No patients in this study had clinically significant adrenal insufficiency and one patient had hypercalcemia (13).
In disseminated TB, seeding of the various endocrine glands with mycobacteria and formation of tubercules is common. In an autopsy study performed in over 100 patients who succumbed to disseminated TB done in the eighties, 53% had involvement of the adrenals, 14% had seeding into the thyroid gland, 5% had direct involvement of the testes, and 4% had seeding into the pituitary gland. Among these 100 patients only one had antemortem clinical adrenal insufficiency (14).
The full spectrum of possible endocrine abnormalities seen with tuberculosis is summarized in Table 1.
Table 1.
Endocrine Abnormalities Seen with Mycobacterium Tuberculosis Infection and with Anti-Tubercular Therapy
View in own window
| Hypothalamus | Diabetes Insipidus |
| Pituitary | Sellar mass lesion
Tuberculous abscess
Sellar Tuberculoma
Thickened stalk with pituitary interruption syndrome
Isolated hyperprolactinemia
Incidental partial or complete hypopituitarism
Isolated hypogonadotropic hypogonadism
Pituitary dysfunction seen with Tuberculous meningitis |
| Thyroid | Tubercular thyroiditis
Cold abscess of the thyroid
Chronic fibrosing thyroiditis
Sick euthyroid syndrome
Para-amino-salicylic acid (PAS) related goiter
Ethionamide and rifampicin related thyroid dysfunction |
| Parathyroid | Inflammation |
| Pancreas | Stress hyperglycemia
Frank diabetes
Pancreatic abscess |
| Testes | Isolated TB orchitis
TB epididymitis
Epididymo-orchitis
Primary gonadal failure |
| Ovaries | Tubo-ovarian abscess
Tubal blockage
Unexplained infertility |
| Water Metabolism | Hyponatremia
Syndrome of inappropriate anti-diuresis (SIAD)
Cerebral salt wasting (CSW) |
| Vitamin D-Calcium Metabolism | Parathyroid hormone independent hypercalcemia
Vitamin D deficiency/hypocalcemia related to isoniazid and rifampicin |
| Adrenals | Tubercular adrenalitis
Addison’s disease
Reversible adrenal insufficiency
Isolated DHEA deficiency |
In this chapter we will review endocrine dysfunction and endocrinopathies associated with TB infection related to the adrenal, thyroid and pituitary glands. Additionally, functional derangement of sodium, and calcium homeostasis will be covered. Glucose intolerance, diabetes and tuberculosis is a large area of public health and will not covered in this chapter.
ADRENALS AND TUBERCULOSIS
TB can involve both adrenal glands primarily or the involvement may be part of disseminated TB. Both conditions may present with primary adrenal insufficiency (Addison’s Disease). Anti-tuberculous therapy (ATT)-related enzyme induction abnormalities can also lead to adrenal dysfunction and in some cases unmask subclinical adrenal insufficiency. Chronic steroid therapy used in the treatment of some types of tuberculous infection can lead to suppression of HPA axis and secondary adrenal insufficiency. Finally, it is important to remember that pituitary involvement in central nervous system (CNS) TB can sometimes lead to isolated corticotropin deficiency with adrenal insufficiency or it can be part of generalized hypopituitarism
Tuberculosis and Addison’s Disease
Thomas Addison in 1855 first described chronic adrenal failure, or Addison’s disease (AD), due to Mycobacterium tuberculosis infection involving both the adrenal glands. In his paper describing AD, 6/11 patients had tuberculous involvement of the adrenal glands. In 1930, Guttman reported a large series of 566 cases with AD, of which 70% was due to tuberculous adrenalitis (15). In 1956 only 25% of AD was related to TB infection (16). The decreasing incidence of tubercular adrenal failure in Western literature was highlighted in a recent large study of 615 cases of AD from Italy in 2011; in this series only 9% of cases were due to TB (17).
This decline in the number of patients with AD related to TB has not been seen in countries endemic for TB like India and South Africa. In India, tuberculous etiology was found in 47% of patients with AD, and of them 85% had enlargement of one or both adrenal glands on imaging (18). The differences between AD due to TB and those with idiopathic AD is summarized in Table 2. In South Africa, 32% of patients with AD had tubercular etiology (19). The most common cause of AD worldwide, however, is autoimmune adrenalitis.
Table 2.
Differences in Clinical Presentation of Tubercular Addison’s Disease (AD) versus Idiopathic AD (18)
View in own window
| Clinical Features | Tubercular | Idiopathic | p-value |
|---|
| Mean age (in years) | 42 | 35 | NS |
| Durations of symptoms before diagnosis (in months) | 14 | 21 | NS |
| Sex Ratio (M: F) | 10:1 | 14:8 | < 0.05 |
| Presentation as crisis (%) | 40% | 23% | NS |
| Evidence of other autoimmune disease (%) | 10% | 27% | < 0.05 |
| Evidence of extra-adrenal TB (%) | 55% | 9% | < 0.05 |
| Adrenal Cytoplasmic Antibodies (%) | 17% | 50% | < 0.05 |
PATHOPHYSIOLOGY OF TUBERCULAR ADRENALITIS
Adrenal TB develops from hematogenous or lymphatic spread, hence is often associated with extra-adrenal infection. The rich vascularity of the adrenal gland and high levels of local corticosteroids that suppress cell mediated immunity create an ideal microenvironment for the growth of Mycobacterium tuberculosis (20). Adrenal involvement can be found in up to 6% of patients with active TB, however isolated adrenal involvement is seen only in a fourth of these (1.5-3% of cases with tubercular infection) (21, 22). Clinical manifestations of AD appear only after 90% of the adrenal cortices have been compromised (23).
The patterns of adrenal gland involvement in TB are summarized below and in (24):
- 1.
Chronic infection of the adrenal gland, with clinical manifestations of primary adrenal insufficiency appearing years after initial infection. Pathologically these patients have small atrophic fibrous glands with or without calcification.
- 2.
Isolated adrenal gland involvement early in the course of disease usually within 2 years of the primary infection. Pathologically these patients most commonly present with bilateral adrenal enlargement because of mass lesions secondary to production of cold abscesses within the adrenal glands. Milder enlargement can be seen in patients with extensive granulomas within the adrenal gland. Lastly, patients with isolated adrenal tuberculosis may also present with normal sized glands with granulomatous inflammation seen microscopically. Calcifications maybe seen in these cases as well.
- 3.
Secondary adrenal insufficiency due to prolonged steroid therapy in disseminated TB or tubercular involvement of the pituitary or hypothalamus.
- 4.
Subclinical steroid deficiency unmasked by ATT-related enzyme induction.
CLINICAL FEATURES
Adrenal TB can be found in any age, however is more commonly seen in adults. Rare cases have also been described in the pediatric age group (25, 26). Thomas Addison’s first description of AD showed a constellation of symptoms like “general languor and debility, remarkable feebleness of heart’s action, and a peculiar change in the color of the skin.” Classic manifestations of AD in the form of malaise or fatigue, anorexia, weight loss, nausea, vomiting, muscle and joint pain, orthostatic hypotension, skin hyperpigmentation and salt craving are often present. Mineralocorticoid deficiency leads to postural hypotension, while hyperpigmentation occurs due to activation of the melanocortin 1 receptors (MC1R) in turn because of high ACTH levels (27). In some patients, however, hyperpigmentation can be absent due to reduced stimulation of MC1R from adrenocorticotropin hormone (ACTH), resulting in an alabaster-like appearance (27). A prior history of TB may also be provided in some patients.
RADIOLOGIC FINDINGS
Computed tomography (CT), magnetic resonance imaging (MRI) and positron emission tomography (PET) are useful non-invasive tools in the diagnosis of adrenal TB. CT has been regarded as the modality of choice for diagnosing adrenal TB, and should include both non-contrast and contrast-enhanced techniques. Adrenal involvement is usually bilateral (28), and findings vary according to course of disease.
Early stage: In the first two years, CT shows noncalcified enlarged adrenals with areas of lucency reflecting caseous necrosis, and a peripheral rim of contrast-enhanced parenchyma. Contours of the adrenal glands are generally preserved.
Late stage: As disease progresses, the adrenals normalize in size, then shrink, and have calcific foci with irregular margins. Calcifications are best visualized on non-contrast CT scans. These may be either diffuse, focal or punctate in nature (
22). These findings correlate with long-standing fibrosis and dystrophic calcification seen with tuberculous granulomas.
LABORATORY FINDINGS
Common findings in patients with adrenal TB include hyponatremia, hyperkalemia and normochromic anemia (27). Hyponatremia can occur due to decreased inhibitory control of vasopressin secretion, resulting in mild SIAD (29). The Mantoux (tuberculin) test usually is strongly positive and erythrocyte sedimentation rate (ESR) is elevated.
In primary AD, baseline serum ACTH levels are higher than 100 pg/ml, plasma renin levels are elevated and serum aldosterone levels are low. In secondary AD, ACTH levels will be low or inappropriately normal, while mineralocorticoid secretion will be normal (
27).
Serum DHEAS will also be low in patients with both primary and secondary AD (
27).
Adrenal insufficiency can be demonstrated by low morning plasma cortisol with a reduced response to synthetic ACTH (
27). Impaired ACTH-stimulated cortisol responses have also been observed with lower baseline cortisol levels, and both higher and lower cortisol responses to ACTH stimulation in patients with isolated pulmonary TB without adrenal involvement (
24). Injectable tetracosactide hexa-acetate, ACTH 1-24 (Synacthen®) (SST), is not marketed or easily available in many developing countries in the world including India. An alternative ACTH which is injectable long-acting porcine sequence, ACTH 1-39 (Acton Prolongatum®) (APST), is easily available and much cheaper. In a study done recently in India by Nair et al in 20 patients with established adrenal insufficiency and 27 controls, the area under the curve of APST (at 120 min) was 0.986 when compared to the standard SST, thus proving its high accuracy. A serum cortisol cut off value of 19.5 µg/dL at 120-min following APST showed a sensitivity of 100% and specificity of 88% (
30).
PATHOLOGY OF ADRENAL TUBERCULOSIS
Macroscopic involvement of the adrenal gland can be seen in up to 46% of patients with adrenal TB. Bilateral involvement is seen in nearly 70% of patients, however they may not be equally affected. Mean combined weight of adrenal gland ranges from 10-37 gm (mean 17 gm) (31). Caseous necrosis can be seen grossly within a large cavity or within multiple scattered tubercles.
Histopathologically, destruction of both the cortex and the medulla is seen with the following patterns of adrenal gland involvement (24):
- 1.
Presence of granulomas, with or without necrosis. The granulomas show epithelioid cell collections with typical Langhan’s giant cells and an admixture of lymphocytes and plasma cells. Ziehl-Neelsen stain is very useful for detecting acid fast bacilli (AFB) within the necrotic areas, as well as within the granulomas.
- 2.
Glandular enlargement with destruction of parenchyma by necrotizing granulomas.
- 3.
Mass lesion secondary to formation of cold abscesses. In these cases, CT-guided fine needle aspiration cytology (FNAC) is helpful for demonstration of AFB, polymerase chain reaction (PCR), and culture for Mycobacterium tuberculosis organisms. In most cases, a combination of histopathology, PCR and culture may be required to confirm the diagnosis.
- 4.
Adrenal atrophy secondary to fibrosis resulting from long-standing tuberculous infection (32).
DIFFERENTIAL DIAGNOSIS
The differential diagnosis for adrenal enlargement includes primary or metastatic tumors, lymphomas, fungal infections like cryptococcus and histoplasma, amyloidosis, sarcoidosis, hemangiomas and adrenal cortical hyperplasia (24, 28). Tissue sampling for microbiological (PCR and culture) and pathological analysis should adequately distinguish between them.
TREATMENT
Treatment for active adrenal TB is similar to the regimen followed for extrapulmonary TB with use of multidrug ATT. Rifampicin induces hepatic enzymes that increase the metabolism of glucocorticoids; hence higher doses of replacement glucocorticoids may be required. Rarely, Rifampicin may trigger an adrenal crisis.
In cases of chronic disease, adrenal gland function is unlikely to recover due to massive destruction of the gland (20, 28). However, a few authors report improvement in adrenal function when patients are given ATT early in the course of disease (33-35). This may be in part due to the remarkable regenerative capacity of the adrenal cortex to undergo hyperplasia and hypertrophy during active infection (20).
Hormone replacement for primary and secondary adrenal insufficiency related to TB follow the same principles as autoimmune or idiopathic primary AD or secondary adrenal insufficiency. In addition to appropriate glucocorticoid replacement mineralocorticoid replacement may be required. Care must be taken to educate patients about stress dosing and the need for parenteral steroids when the patient may not be able to take or absorb oral glucocorticoids.
THYROID AND TUBERCULOSIS
The thyroid gland is an uncommon site for infection by M. tuberculosis. Thyroid TB (TTB) is therefore very rare, even in places with a high prevalence of TB. The primary presentation of TTB is as a mass or a goiter. Overt hormonal dysfunction is very uncommon in TTB. However, in patients with tuberculosis affecting any organ clinically insignificant abnormalities in thyroid function tests are very common. ATT also causes both structural and functional thyroid dysfunction. Pre-operative diagnosis of TTB can be made only with a high index of suspicion while evaluating thyroid nodules especially in communities with a high prevalence of TB (36).
Epidemiology
M. tuberculosis has been documented to be involved in the thyroid gland of 0.1 to 1% of patients who underwent thyroid tissue sampling for any indication (37-39). In an autopsy series of patients with advanced disseminated TB occurring in the pre- and post-antibiotic era, 14% had evidence of thyroid gland involvement (14). In a large cohort of 2,426 patients from Morocco, only eight had evidence of TB (0.32%). These were in the form of goiter or as a solitary thyroid nodule. In a study from India, thyroid involvement has been seen in 0.43% of specimens obtained from FNAC (40), while among Turkish patients undergoing thyroidectomy, 0.25 - 0.6% showed thyroid involvement by M. tuberculosis (41, 42).
Pathogenesis
Thyroid involvement in TB is very uncommon. A few postulated intrinsic properties of the thyroid which are proposed not to allow Mycobacterium tuberculosis bacilli to survive include (24, 36):
Presence of iodine-containing colloid possessing bacteriostatic activity.
High blood flow within the thyroid gland with the presence of intracellular iodine.
Increased phagocytosis within the gland, seen in hyperthyroidism.
Rich lymphatic supply to the thyroid.
Thyroid hormones themselves exercise anti-TB roles.
TTB can be primary or secondary
- A.
Primary TTB is involvement of the thyroid gland alone, with no evidence of TB elsewhere in the body.
- B.
Secondary TTB is usually the result of hematogenous, lymphatic and/or direct spread from an active tubercular focus involving the cervical lymph nodes or larynx. Secondary TTB is much more commonly encountered than primary TTB, and TTB may go undiagnosed in many cases especially where clinical signs are non-specific (24).
Clinical Features
TTB occurs slightly more commonly in women as compared to men (M: F = 1:1.4) and occurs over a wide age range of 14 to 83 years, median age of 40 ± 16 years for men and 43 ± 17 years for women (36).
TTB can manifest as a localized swelling with cold abscess mimicking carcinoma, as multinodular goiter, as a solitary thyroid nodule without cystic component, or very rarely as an acute abscess. The various presentations are summarized in . Rarely TTB may present as a goiter or a chronic fibrosing thyroiditis. Presence of cervical lymphadenopathy may raise suspicion of malignancy (36). Clinical presentation is often subacute, but may be acute in cases of abscess (43). Pain associated with swelling, thyroid tenderness, fever and localized extra-thyroidal findings such as dysphagia, dysphonia or recurrent laryngeal nerve palsy are less common in TTB as compared to patients with acute bacterial thyroiditis (24). However, some patients with TTB may present with pyrexia of unknown origin. Table 3 documents with differences in clinical presentation between TTB and bacterial thyroiditis.
Table 3. Clinical Features that Help Differentiate Between Tuberculous Thyroiditis and Bacterial Thyroiditis
View in own window
| Clinical Features | Tuberculous Thyroiditis | Bacterial Thyroiditis |
|---|
| Pain | - | +++ |
| Pyrexia | +1 | +++ |
| Duration of illness (mean duration) (Ref;24) | 105 days | 18 days |
| Dysphagia | ++ | +++ |
| Dysphonia | ++ | +++ |
| Recurrent Laryngeal Nerve Palsy (Hoarseness) | ++ | +++ |
| History of previous thyroid illness | - | + |
| Tenderness over the gland | - | +++ |
| Leukocytosis | - | ++ |
| Elevated Erythrocyte Sedimentation Rate (ESR) | +++ | + |
1Rare reports of presentation as pyrexia of unknown origin
Most patients with TTB are euthyroid and do not have pre-existing thyroid disease. Very rarely TTB can be associated with hypothyroidism, with a period of subclinical hyperthyroidism preceding the hypothyroidism (44). Myxedema can occur in cases with extensive destruction of the thyroid gland by disseminated TB, which can also be fatal (45). Past history of TB may be elicited in some cases, and patients may have history of cervical lymphadenopathy (43).
Radiological and Laboratory Findings
Chest X-ray, ESR, and tuberculin skin test should be performed in all cases of suspected TTB. The diagnosis is made only after FNAC or histopathological examination of the surgical specimen when FNAC is negative (43). Sputum AFB may rarely assist in diagnosis in cases with associated pulmonary TB.
Ultrasonography usually shows a heterogenous, hypoechoic mass similar to a neoplastic nodule. Anechoic areas with internal echoes may be seen in abscesses. Contrast-enhanced CT scan can determine the location of the necrotic lesions (46).
On MRI the normal thyroid is homogenously hyperintense relative to the neck muscles on both T1 and T2-weighted images. TTB may show intermediate signal intensity due to granulomatous inflammation, however, this appearance is also seen in thyroid carcinoma. Abscesses appear hypointense on T1 and hyperintense on T2-weighted images, and may show peripheral rim of contrast enhancement (47).
Thyroid function tests (TFT) are usually normal in patients with TTB. Thyrotoxicosis in the initial stage of rapid release of thyroid hormone, and myxedema in the later stage of thyroid gland destruction have also been noted, and patients may have abnormal TFT accordingly (24). Only 5.2% of patients with TTB have abnormal TFT (36).
Pathology of Thyroid TB
In most cases, TTB can be diagnosed on FNAC which typically shows epithelioid cell granulomas with Langhan’s giant cells, peripheral lymphocytic infiltration and purulent caseous necrosis. The yield of AFB by the Zeihl Neelsen stain is more with FNAC samples than in biopsies. The aspirates can be sent for TB culture or PCR. TB-PCR is much more sensitive in detecting M. tuberculosis deoxyribonucleic acid (DNA) from FNA samples, and is an alternative to rapid diagnosis of TB in AFB-negative cases (40). The diagnosis is substantiated by histopathology which typically shows granulomas, Langhan’s giant cells and necrosis (). Few cases show dense lymphocytic infiltrate with prominent germinal centers, resembling lymphocytic or Hashimoto thyroiditis ().
Five pathological varieties of TTB have been described (36):
- 1.
Multiple miliary lesions throughout the thyroid gland
- 2.
Goiter with caseation necrosis
- 3.
Cold abscess
- 4.
Chronic fibrosing tuberculosis
- 5.
Acute abscess
Differential Diagnosis
TTB, although rare, should be considered in the list of differentials for solitary or multinodular thyroid nodules, and abscesses (36). Reidel’s thyroiditis may mimic chronic fibrosing tuberculosis clinically, however histopathology clinches the diagnosis (42).
Treatment
ATT remains the cornerstone of treatment. Surgery has a limited role with drainage of abscess, avoiding total destruction of gland and subsequent hypothyroidism. However inadvertent total thyroidectomies are performed as the pre-operative diagnosis is commonly a malignancy. In cases were TB was diagnosed prior to surgery, ATT is well tolerated with resolution of symptoms, reduction in thyroid mass symptoms, and with favorable reversal of thyroid hormonal dysfunction. Standard ATT schedules are followed. Thyroid hormone levels should be monitored before, during, and after treatment. Despite strict ATT, recurrence and failure rate is 1% due to resistance to ATT drugs (48).
Functional and Structural Alterations of Thyroid Functions with Active Tuberculosis and with Anti-tubercular Therapy
Among hospitalized patients with TB without any evidence of involvement of the thyroid gland sick euthyroid syndrome with low free T3 is common. The estimates vary between 63-92% and probably is the commonest endocrinopathy seen in patients with TB (13,49). As with other unwell patients the degree of reduction in Free T3 serves both as a marker for severity of the disease and mortality. In the study by Chow et al, all patients who survived the hospitalization had normal TFT within one month of initiation of ATT. In community dwelling patients with TB, the prevalence of thyroid dysfunction is unclear.
Thyroid hormones are metabolized in the liver and the kidneys. In the liver, the enzyme CYP3A4 belonging to the hepatic cytochrome P450 family is responsible for the metabolism. Rifampicin is a potent activator of the P450 system and this leads to an increase in T4 turnover. In most adults with normal a hypothalmo-pituitary-thyroid axis this increase in turnover is compensated by an increase in the production of thyroid hormones and a slight increase in thyroid volume. This may be noted biochemically as a slight increase in free T3 and total T3 levels after rifampicin administration. There are no changes in free T4 and TSH concentrations (50). Among patients with pre-existing thyroid disease with a limited capacity to increase production of thyroid hormones, the rifampicin-mediated increase in free T4 turnover might lead to the need for an increase in thyroid hormone replacement therapy. In a retrospective cohort of patients on levothyroxine replacement therapy, the addition of rifampicin as part of ATT led to a need for a 26% increase in dose in patients on thyroid hormone replacement therapy and a 50% increase in patients on suppressive therapy post thyroidectomy for differentiated thyroid cancer (51).
Older anti-tubercular agents have more profound effects on thyroid physiology. Studies by Munkner et al demonstrated an association between the use of p-amino salicylic acid (PAS) and the development of goiter (52). PAS and ethionamide were also associated with significant risk of developing hypothyroidism (53, 54). However, these agents are currently not used as first line agents. It is prudent to monitor TFTs 6-8 weeks after initiation of any of these three agents in patients who have pre-existing thyroid dysfunction.
PITUITARY AND TUBERCULOSIS
Direct involvement of the pituitary gland by Mycobacterium Tuberculosis is very rare. Some of the earliest published reports of pituitary TB include von Rokitansky who noted tubercles in the hypophysis as early as 1844, Letchworth in 1924 who reported a case of primary pituitary tuberculoma on autopsy examination, and Coleman and Meredith documented a case of pituitary TB in 1940 (55, 56).
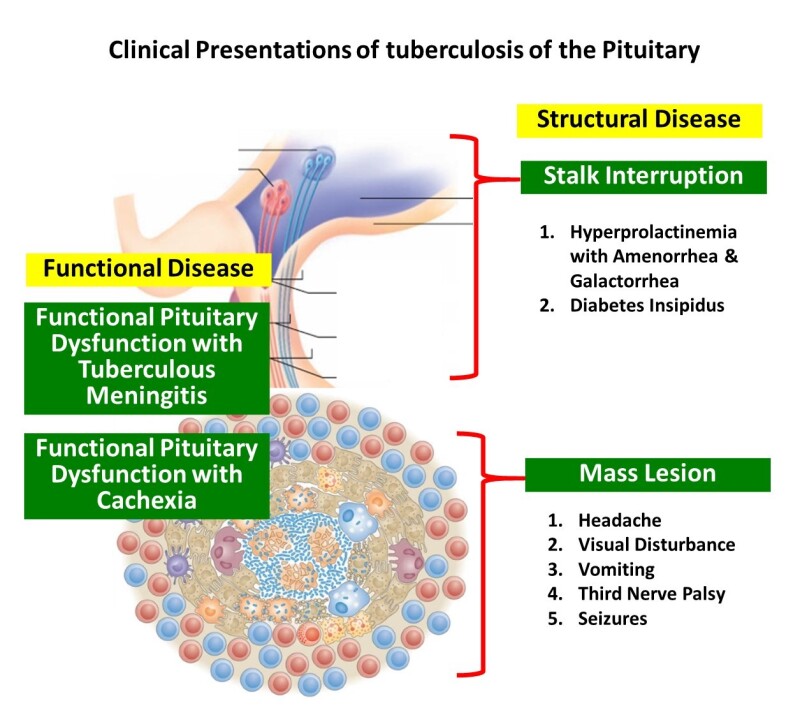
The spectrum of involvement () of the pituitary gland with TB includes sellar, parasellar, and stalk tuberculomas and sellar tubercular abscesses. Patients with tuberculous meningitis exhibit a range of functional pituitary dysfunction even in the absence of any evidence of direct invasion/extension of the disease into the sella. Among survivors of tubercular meningitis hypopituitarism was noted 10 years after the primary disease. Hypothalamic pituitary dysfunction such as isolated hypogonadotropic hypogonadism may accompany cachexia and weight loss that can complicate more extensive disease. Infiltrative tubercular disease of the stalk can produce pituitary interruption syndrome including isolated diabetes insipidus and hyperprolactinemia.
In most cases the diagnosis of tuberculosis of the pituitary is established on histopathology, often in the absence of confirmatory culture studies or positive acid-fast stains (57). Although the diagnosis is difficult on clinical and radiological examination, pituitary TB should be considered in the differential of a suprasellar mass especially in developing countries, as the condition is potentially curable with ATT (58, 59).
Sellar Tuberculoma/Abscess
EPIDEMIOLOGY
The incidence of pituitary TB is very low. In an autopsy series of 3,533 cases, only 2 of 89 intracranial tuberculomas involved the sella turcica, while in another autopsy series of 14,160 cases, only 2 cases of TB were encountered involving the anterior pituitary lobe (50). In patients with late generalized TB, the incidence of pituitary involvement is 4% (14). Nearly 70% of pituitary TB reported worldwide has been reported from the Indian subcontinent, probably attributable to the higher prevalence of TB in this location (60). In the largest series from India, 18 cases of sellar TB were diagnosed based on histopathology from 1148 pituitary surgeries (60).
PATHOGENESIS
Pituitary TB can arise either from hematogenous seeding, in the presence or absence of miliary disease, or from direct extension from the brain, meninges or sinuses. TB can either involve the pituitary gland alone, or involve the adjacent and/or distant organs as well (60). Both the adenohypophysis and neurohypophysis may be involved by TB. Supra-sellar extension is common in pituitary TB with only rare cases confined to the sella (57).
CLINICAL FEATURES
Pituitary TB occurs at a mean age of 34.1 ± 13.6 years (age range 6 to 68 years), and is more common in women (F:M = 2.7:1). Young children are at high risk of progression of TB including CNS disease. Clinical presentation is often indolent. Duration of symptoms average 4 months (60, 61).
Pituitary involvement, either as a sellar abscess or tuberculoma presents primarily with symptoms of a sellar mass. The common presentations clinically are gradual onset of headache (85.2%), visual loss (48.1%) (), seizures and cranial nerve palsies. Patients with infiltration of the stalk by tuberculomas may present with central diabetes insipidus with polyuria (8.6%) or menstrual abnormalities related to hyperprolactinemia like amenorrhea in women (37.3%) and galactorrhea (23.7%) (60, 61). Growth retardation and hypogonadism are rare findings in children with pituitary TB (61). Hyperphagia resulting in obesity or weight gain has also rarely been documented which may occur due to the loss of sensitivity of the appetite-regulating network in the hypothalamus to afferent peripheral humoral signals (62). Apoplexy, characterized by acute infarction and/or hemorrhage in the pituitary gland, is an uncommon presentation of pituitary TB (63). Systemic and constitutional symptoms may or may not be present; low grade fever may be seen in 14.8% of patients. Other organs may show evidence of TB in 26.9% (60, 64). Tuberculous meningitis may be associated in a few cases.
RADIOLOGICAL FINDINGS
The diagnosis of primary pituitary TB is challenging and often difficult. Radiologically pituitary TB can mimic pituitary adenoma, arachnoid cyst, pyogenic abscess, metastasis, or craniopharyngioma. MRI typically shows a sellar mass which may extend into the suprasellar region, involving the optic nerves and inter-carotid space (). T1-weighted MR images appear isointense. T2-weighted images show central hyperintensity corresponding to caseous necrosis, and gadolinium contrast imaging may show thick ring enhancement in the periphery with central hypointense areas. Meningeal enhancement with enhancement of the thickened pituitary stalk may favor non-adenoma etiology. Additional findings like sellar/suprasellar calcification and sellar floor erosion have also been described (57, 63-65).
MR spectroscopy can detect elevated lipid peaks in a tuberculoma at 0.9, 1.3, 2.0 and 2.8 ppm, and a phosphoserine peak at 3.7 ppm. Lipid resonance at 0.9 and 1.3 ppm occur due to methylene and terminal methyl groups on fatty acids found in caseous necrosis (66)
LABORATORY FINDINGS
Panhypopituitarism may be encountered on evaluation of anterior pituitary hormones like thyroid stimulating hormone (TSH), early morning cortisol, growth hormone, prolactin, luteinizing hormone (LH) and follicle-stimulating hormone (FSH).
Testing for HIV and immunocompromised states should be considered in the appropriate clinical setting. Positive tuberculin test and elevated ESR may be seen in patients with systemic involvement.
PATHOLOGY OF PITUITARY TUBERCULOSIS
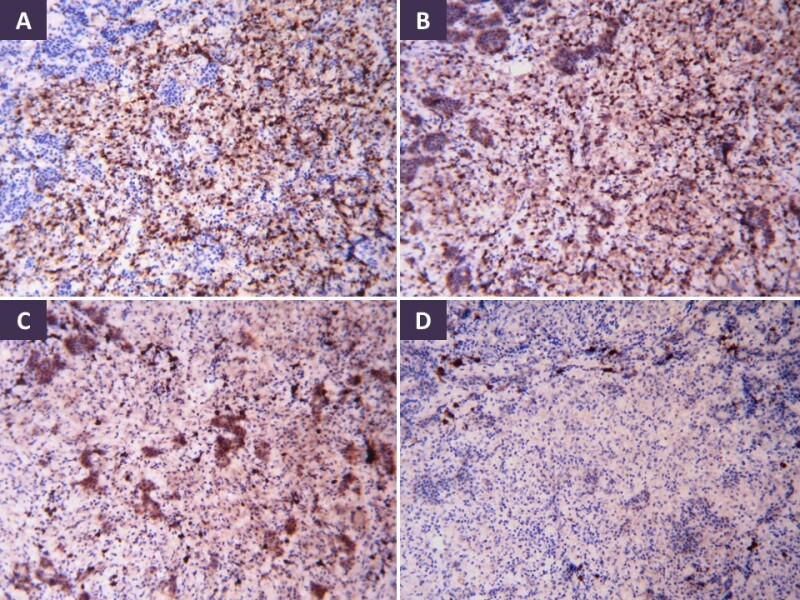
The most common pattern of tuberculous involvement of the pituitary on histopathological examination is granulomas without caseation necrosis (59.6%) as show in . Granulomas may be miliary or may coalesce together to form a conglomerate mass. Reticulin stain helps demonstrate loss of normal reticulin pattern of the pituitary (). The granulomas are composed of epithelioid cells, Langhan’s giant cells, and lymphocytes (). Immunohistochemistry (IHC) with CD68 can help confirm the presence of epithelioid histiocytes in cases of unequivocal morphology, while CD3, CD20 and CD138 can highlight a mixture of T-lymphocytes, B-lymphocytes and plasma cells, respectively (). Pus and caseation necrosis are seen less commonly, and in these cases the yield of AFB is greater than with cases without necrosis. As with other sites, demonstration of AFB on biopsy material is very low. In such cases growth of M. Tuberculosis organisms on culture and TB PCR aid in diagnosis.
DIFFERENTIAL DIAGNOSIS
Sarcoidosis must be considered in the differential of non-caseating granulomatous hypophysitis, and shows naked granulomas without infiltrating lymphocytes. IgG4-related disease typically shows increase in plasma cells of the IgG4 subtype with storiform fibrosis. Histiocytic lesions like Langerhans cell histiocytosis (LCH) usually involve the infundibulum and typically show presence of Langerhans’s histiocytes with eosinophils. LCH is more common in children and young adults. Other inflammatory lesions involving the pituitary stalk are lymphocytic infundibuloneurohypophysitis (LINH), Wegener’s granulomatosis, and pituitary stalk parasitosis (67). Fungal granulomas involving the hypophyseal region can be ruled out by performing fungal stains on tissue sections (60).
TREATMENT
Transsphenoidal approach is preferred for surgery and is used for diagnosis and decompression of adjacent structures. Typical intra-operative findings are firm to hard, non-suckable greyish tissue with thickening of the dura. Pituitary TB can be managed conservatively if the diagnosis is confirmed with cerebrospinal fluid TB PCR and other tests.
ATT may be given for up to 18 months and patients should be on periodic follow up with assessment of hormonal profile. Lifelong replacement of hormones may be required in some patients (68). Recurrence of TB in lymph nodes despite completion of 18 months of ATT has been reported to occur due to resistance of M.tuberculosis bacilli to Rifampicin (69).
Pituitary Dysfunction in Patients with Tuberculous Meningitis (TBM)
In an Indian study of 75 patients with tuberculous meningitis, common pituitary functional abnormalities included hyperprolactinemia (49%), cortisol insufficiency (43%), central hypothyroidism (31%) and multiple hormone deficiencies (29%) (70). Prevalence of functional pituitary abnormalities seen in TBM in multiple studies from India is summarized in Table 4 (71, 72). In addition, there may be hyponatremia.
Table 4.
Pituitary Involvement in Patients with Tuberculous Meningitis
View in own window
| Delhi (70) | Chandigarh (71) | Lucknow (72) |
|---|
| Number of patients | 75 | 63 | 115 |
| Any Involvement of Pituitary | | 84.2% | 53.9% |
| Single Axis Involvement | | 39.8% | 30.4% |
| More than one axis (Panhypopituitarism) | 29.3% | 44.4% | 23.5% |
| Hypogonadotropic Hypogonadism | NR | 38.1% | 33.9% |
| Hyperprolactinemia | 49.3% | 49.2% | 22.6% |
| Secondary Adrenal insufficiency | 42.7% | 42.9% | 13% |
| Central hypothyroidism | 30.7% | 9.5% | 17.4% |
| Isolated Growth hormone deficiency | NR | NR | 7.8% |
| Syndrome of Inappropriate anti-diuresis | NR | NR | 9.% |
| Diabetes Insipidus | Nil | Nil | Nil |
Even among patients who survive tuberculous meningitis, pituitary dysfunction may persist. A study done by Lam in Hong Kong showed growth hormone deficiency to be the most common finding in patients younger than 21 years of age with tuberculous meningitis after 10 years of surviving tuberculous meningitis (73).
WATER IMBALANCE AND TUBERCULOSIS
Hyponatremia has been commonly seen in patients admitted to the hospital with TB. Though data about the prevalence of hyponatremia among community treated patients with uncomplicated pulmonary TB is sparse, among inpatients admitted with TB, hyponatremia has been seen in 10-76% of patients (74-78). The commonest cause of hyponatremia is the SIAD. Other causes include untreated primary or secondary adrenal insufficiency, volume depletion, hyponatremia associated with volume excess, and hypoalbuminemia and rare cases of cerebral salt wasting seen with tuberculous meningitis (79) (). Hypernatremia is rarely encountered and usually signifies involvement of the hypothalamus or the pituitary stalk leading to diabetes insipidus.
Syndrome of Inappropriate Antidiuresis (SIAD)
In the absence of adrenal deficiency, patients with non-CNS TB who are adequately hydrated (euvolemic) the hyponatremia is almost always a consequence of retention of free water despite low serum osmolality (inappropriate antidiuresis). Wiess and Katz first noted the association between active untreated TB and syndrome of inappropriate antidiuresis (SAID). In four patients with active TB and hyponatremia they noted excessive urinary sodium excretion. When these four patients were put on fluid restriction there was an improvement in the serum sodium levels. All patients who survived also had gradual normalization of serum sodium levels and SAID with treatment of TB (80).
Three different mechanisms have been proposed for the development of SIAD in patients with tuberculosis without evidence of adrenal involvement.
- 1.
The first proposed mechanism in common with other pulmonary diseases is the stimulation of baroreceptors by chronic hypoxemia that can accompany extensive pulmonary TB. There is release of anti-diuretic hormone (ADH) in response to baroreceptor stimulation which leads on to SIAD (81).
- 2.
The second possible mechanism proposed is a shift of the “osmostat” towards the left as seen in patients with decreased effective circulating volume leading to ADH release at lower serum osmolality. Investigators have noted higher circulating ADH levels in the serum despite hyponatremia which subsequently declined when free water was administered. The intact response to hypoosmolality suggested that the osmoregulation set up in the hypothalamus was functioning normally but at a lower osmolar threshold for ADH release (82).
- 3.
The third mechanism proposed is the ectopic secretion of ADH by the tubercular granuloma. This mechanism was proposed by the authors of a case where a patient with well-established diabetes insipidus developed SIAD and hyponatremia after contracting pulmonary tuberculosis (83).
Patients with CNS TB have a higher prevalence of hyponatremia compared to those with pulmonary infections. In adult patients with TBM the prevalence of a low sodium state has varied from 45-65% in different studies (84-86). In children with TBM the prevalence varied from 38-71% in different studies (87-89). In children with TBM and hyponatremia there appears to be an association with mortality and increased intracranial pressures (87, 90). A recent review from India compiled data from over 11 studies comprising a total of 642 patients with TBM and found the prevalence of hyponatremia to be 44%. Unlike non-CNS TB the commonest etiology of hyponatremia among patients with CNS TB is cerebral salt wasting (CSW) rather than SIAD (36% vs 26%) (86). The other less common causes of hyponatremia encountered in TBM include the following
- 1.
Dehydration and hypovolemic hyponatremia due to anorexia, vomiting, nausea and diarrhea
- 2.
Drug induced including use of diuretics, osmotic agents like mannitol and anti-seizure medications like carbamazepine and phenytoin.
- 3.
Secondary adrenal insufficiency and rarely primary adrenal insufficiency
CLINICAL PRESENTATION AND TREATMENT OF SIAD ASSOCIATED WITH TUBERCULOSIS
Most patients with SIAD and TB are asymptomatic and do not require any treatment. The hyponatremia accompanying SIAD self corrects itself when ATT is started (82). Fluid restriction is only required in symptomatic patients or in patients with severe hyponatremia. Prior to restricting fluids in patients with non-CNS TB it is important to rule out dehydration either by assessing volume status clinically, assessing volume status with urine spot sodium levels, or measuring central venous pressures in unwell patients. In patients with CNS TB, it is important to rule out CSW prior to initiating fluid restriction. Hypertonic saline infusions are limited to patients with life threatening symptoms like seizures and deep coma attributable to hyponatremia (86). Care should be taken to correct hyponatremia at a rate not faster than 8-10 mEq/L in 24 hours to avoid central pontine myelinolysis.
Cerebral Salt Wasting (CSW)
CSW refers to changes in renal salt handling that accompanies CNS disorders which leads to natriuresis and hypovolemia. The accompanying dehydration and decrease in effective circulating volume triggers ADH release via baroreceptors. The action of ADH on collecting tubules then leads to selective water resorption and relative water excess and hyponatremia despite overall hypovolemia. The putative renal natriuretic triggers include atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and c-type natriuretic peptide. In TBM the most likely natriuretic trigger is BNP (91). What induces BNP release in patients with TBM is less well characterized. The putative mechanisms that trigger BNP release include sympathoadrenal activation, increase in intracranial pressure, and vasospasm in cerebral arteries (92). In patients with CNS TB statistically CSW is more likely to be the cause of hyponatremia and should always be ruled out prior to labelling them as SIAD and starting fluid restriction. A simple diagnostic criterion proposed by Kalita et al (86) includes meeting all the 3 essential criteria and meeting at least 3 out of 5 additional supportive criteria to label the patient has having CSW. Table 5 lists the clinical and biochemical differences between SIAD and CSW.
Essential Criteria (meets all three)
- 1.
Polyuria (>3 liters of urine in 24 hours over 2 days)
- 2.
Documented hyponatremia
- 3.
Exclusion of other cause of natriuresis like adrenal insufficiency, salt losing nephropathies, use of diuretics, hepatic and cardiac failure.
Additional criteria (meet 3 out of 5)
- 1.
Clinical evidence of hypovolemia and dehydration
- 2.
Documented negative fluid balance either by careful weight monitoring or by strict intake and output records
- 3.
Urine spot Sodium > 40 mEq/L
- 4.
Central Venous Pressure (CVP) < 6 cm of water
- 5.
Laboratory evidence of dehydration including an increase in hemoglobin and hematocrit, increase in blood urea nitrogen and increase in albumin than previously.
Table 5.
Differentiation Between Syndrome of Inappropriate Anti-Diuresis (SIAD) and Cerebral Salt Wasting (CSW)
View in own window
| Parameter | SIAD | CSW |
|---|
| Extracellular Volume | Increased | Decreased |
| Body Weight | Increased | Decreased |
| Fluid Balance | Positive | Negative |
| Tachycardia | - | + |
| Hypotension | - | + |
| Hematocrit/Albumin/Blood Urea Nitrogen | Normal | Increased |
| Central Venous Pressure | Normal or slightly high | Decreased |
TREATMENT OF CSW ASSOCIATED WITH TBM
The primary treatment for CSW is fluid replacement with or without oral salt loading for as long as polyuria continues. Isotonic fluids are preferred for replacement. If the patient has a central venous line then the central venous pressure (CVP) measurements would guide the fluid replacements. In the absence of a CVP line fluid balance is needed by either meticulous intake and output charting or use of daily weight measurements.
In patient’s refractory to fluid replacement and oral salt loading, oral fludrocortisone (OFC) has been tried as there is an inhibition of the renin-angiotensin-aldosterone system (RAAS) system in CSW. A recent randomized control trial was conducted in 36 patients with CSW associated with TBM. Half of them received OFC (0.4-1mg/day) plus fluid and oral salt and the other half received only fluids and oral salt. The patients who received OFC in addition had quicker normalization of serum sodium levels (4 days vs 15 days; p 0.04) and lesser cerebral infarctions related to vasospasm (6% vs 33%; p 0.04). However, OFC use was associated with severe hypokalemia and significant hypertension in 2 patients each and in one patient there was an episode of pulmonary edema. OFC had to be withdrawn in 2/18 patients because of these serious adverse events. There was no difference in mortality or disability at 3 and 6 months among patients who received OFC vs the patients who did not (93).
CALCIUM ABNORMALITIES IN TUBERCULOSIS
Hypercalcemia in Patients with Tuberculosis
Hypercalcemia has been known to be associated with a number of granulomatous diseases. The three commonest granulomatous diseases causing hypercalcemia include sarcoidosis, TB, and fungal infection (94). The prevalence of hypercalcemia in patients with TB has ranged from 2-51% in studies done from South Africa (2%), Hong Kong (6%), India (10.6%), Sweden (25%), Malaysia (27.5%), Greece (25% & 48%) and Australia (51%) (13, 95-101). In contrast, prospective studies from the United Kingdom, Belgium, and Turkey did not show any hypercalcemia among patients with newly diagnosed tuberculosis (102-104). The primary determinant in the development of hypercalcemia among patients with TB appears to be their Vitamin D status and nutritional calcium intake. In populations with high nutritional calcium intake and adequate sunlight exposure like in Greece and Australia the prevalence of hypercalcemia is highest. Among countries with good sunlight exposure but poor nutritional calcium intake like most Asian countries there is a more modest prevalence of hypercalcemia. The countries with good nutritional calcium intake but poor sunlight exposure and low Vitamin D levels appear to have the lowest prevalence of hypercalcemia. This has been elegantly explained by Chan et al (105). However, some outliers like the higher prevalence in Sweden and moderate prevalence in India are not completely explained by this hypothesis alone.
In a recent paper looking at retrospective records of patients admitted with TB at a tertiary care hospital in Vellore almost 20% of patients were found to have albumin-adjusted hypercalcemia. The authors looked at the risk factors for hypercalcemia by comparing them with the patients without hypercalcemia assuming that background nutritional calcium intake and Vitamin D levels were similar. The primary risk factors for the development of hypercalcemia within this group was presence of renal dysfunction or frank renal failure, use of diuretics, disseminated tuberculosis, and presence of co-morbidities like diabetes and hypertension (106).
MECHANISM OF HYPERCALCEMIA WITH TUBERCULOSIS
The definitive mechanism that causes hypercalcemia among patients with TB is still unclear. Alternative etiologies for hypercalcemia including adrenal insufficiency, primary hyperparathyroidism, primary hyperthyroidism, milk alkali syndrome have been ruled out in many of the case series. Biochemically several investigators have shown an increase in the levels of 1,25-dihydroxy vitamin D along with low or normal levels of 25-hydroxy vitamin D levels. This suggests an increase in the conversion of 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D (107). This conversion is mediated by the enzyme 1-α-hydroxylase found in the kidney. However, hypercalcemia is even reported in patients with chronic renal failure and among those with absent kidneys (108, 109). This suggests a non-renal site of enzymatic activity.
The tubercular granuloma is suggested as the site for the extra renal 1-α-hydroxylase activity (110). Activated macrophages can express 1-α-hydroxylase activity and in patients with active TB activated macrophages retrieved by broncho-alveolar lavage were able to synthesize 1,25 dihydroxy vitamin D in vitro studies (111). The macrophage production of 1-α-hydroxylase is probably important for the immune response to tuberculous infection. The binding of active Vitamin D (1,25-dihydroxy vitamin D) to Vitamin D receptors within the immune cells stimulates autophagy and production of cytokines that contribute to the clearance of the mycobacterium from the body (112, 113). In addition, active Vitamin D contributes to the downregulation of the inflammatory response of the body to reduce damage to bystander host tissues (114). The increased intestinal absorption of calcium and observed hypercalcemia may be an unintended consequence of this immune-protective phenomenon.
This also explains why patients with low levels of substrate (25-hydroxy vitamin D) for the enzyme or among those with poor calcium intake there is less likelihood of the development of hypercalcemia.
CLINICAL PRESENTATION
Most patients with hypercalcemia related to TB infection are asymptomatic. Rarely patients develop symptoms related to hypercalcemia including polyuria, anorexia, nausea, weakness and lethargy, more serious CNS symptoms like delirium.
Patients may develop hypercalcemia later in the course of TB after commencement of therapy with improvements in nutritional and albumin status and improvement in nutritional calcium intake.
TREATMENT
A Cochrane review of Vitamin D supplementation in patients with TB did not show any benefits in terms of improved outcomes but there was also no increased risk of developing hypercalcemia (115). Most patients have gradual resolution of hypercalcemia on ATT over 1 to 7 months (96).
Hypocalcemia in Patients Treated with Rifampicin and Isoniazid
Hypocalcemia was noted for the first time in United Kingdom during a randomized control trial of anti-tubercular chemotherapy after several months of therapy. Fourteen out of the 325 patients on the trial developed hypocalcemia. In this trial none of the 325 patients was noted to have hypercalcemia. On the whole as a group the mean calcium levels dropped significantly during the course of the treatment trial. The mechanism is proposed to be the action of both Rifampicin and Isoniazid on vitamin D metabolism (102).
When isoniazid is given to normal subjects there is a brisk decline in the levels of active Vitamin D (1,25 dihydroxy vitamin D). There is slower decline in the levels of 25-hydroxy vitamin D accompanied by a compensatory increase in the levels of parathyroid hormones. In the same study isoniazid was shown to inhibit cytochrome p450 related hepatic mixed function oxidase and it is assumed that since the renal 1-α Hydroxylase is also related to cytochrome P450 system there would be decreased conversion of 25-hydroxy vitamin D to 1,25 dihydroxy vitamin D (116).
On the other hand, rifampicin is an inducer of hepatic hydroxylase which should in theory lead to an increase in active Vitamin D levels. However, when rifampicin was given to normal volunteers there was a fall in 25-hydroxy vitamin D levels with no changes to the levels of 1,25-dihydroxy vitamin D. The possible explanation for this decline is likely to be that the higher metabolic turnover of active Vitamin D induced by rifampicin is not compensated by an increase in dermal production or increased nutritional provision of vitamin D (117). Regardless, in treatment regimens that include both rifampicin and isoniazid there is a very real possibility of the development of not just hypocalcemia but unmasking of rickets and osteomalacia especially when the patient is poorly nourished.
CONCLUSIONS
TB can involve almost all endocrine glands as a primary disease-causing destruction and loss of function. In enclosed spaces like the pituitary fossa and neck the granuloma/tuberculoma/cold abscess can replace vital structures and cause symptoms related to a mass. This chapter did not cover direct tubercular involvement of the ovaries, testes, and the pancreas.
Additionally, a whole range of functional hormone abnormalities can accompany the effect on chronic inflammation on the immune-endocrine pathways. Metabolic derangement in calcium and water metabolism are covered in detail. Abnormalities in glucose metabolism are not covered because of the vast amount of information now available on the public health aspects of TB and diabetes mellitus.
Fortunately, most abnormalities are self-limited and resolve with successful ATT. However, one needs to consider the rare possibility of a hormonal emergency like an adrenal crisis, hypercalcemic emergency, or pituitary apoplexy in the context of TB.
REFERENCES
- 1.
- 2.
Lyon SM, Rossman MD. Pulmonary tuberculosis. Microbiol Spectr (2017) 5:TNMI7–0032. doi: 10.1128/microbiolspec.TNMI7-0032-2016. [
PubMed: 28185620] [
CrossRef]
- 3.
- 4.
Besedovsky H, del Rey A. Immune-neuro-endocrine interactions: facts and hypothesis.
Endocr Rev. 1996;17:64–95. [
PubMed: 8641224] [
CrossRef]
- 5.
Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action.
Physiol Rev. 1999;79:1–71. [
PubMed: 9922367]
- 6.
Straub RH, Schuld A, Mullington J, Haack M, Schölmerich J, Pollmächer T. The endotoxin-induced increase of cytokines is followed by an increase of cortisol relative to dehydroepiandrosterone (DHEA) in healthy male subjects.
J Endocrinol. 2002;175:467–74. [
PubMed: 12429044]
- 7.
Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NFκB activity through induction of IκB synthesis.
Science. 1995;270:286–90. [
PubMed: 7569976]
- 8.
Pandolfi J, Baz P, Fernández P, Discianni Lupi A, Payaslián F, Billordo LA, et al. Regulatory and effector T-cells are differentially modulated by dexamethasone.
Clin Immunol. 2013;149:400–10. [
PubMed: 24211714]
- 9.
van Lettow M, van der Meer JWM, West CE, van Crevel R, Semba RD. Interleukin-6 and human immunodeficiency virus load, nut not plasma leptin concentration, predict anorexia and wasting in adults with pulmonary tuberculosis in Malawi.
J Clin Endocrinol Metab. 2005;90:4771–6. [
PubMed: 15928249]
- 10.
Kellendonk C, Eiden S, Kretz O, Schütz G, Schmidt I, Tronche F, et al. Inactivation of the GR in the nervous system affects energy accumulation.
Endocrinology. 2002;143:2333–40. [
PubMed: 12021198]
- 11.
Plata-Salaman CR. Central nervous system mechanisms contributing to the cachexia-anorexia syndrome.
Nutrition. 2000;16:1009–12. [
PubMed: 11054608]
- 12.
D’Attilio L, Santucci N, Bongiovanni B, Bay ML, Bottasso O. Tuberculosis, the Disrupted Immune-Endocrine Response and the Potential Thymic Repercussion as a Contributing Factor to Disease Physiopathology.
Front. Endocrinol. 2018;9:214. [
PMC free article: PMC5938357] [
PubMed: 29765355]
- 13.
Post FA, Soule SG, Willcox PA, Levitt NS. The spectrum of endocrine dysfunction in active pulmonary tuberculosis.
Clin Endocrinol (Oxf). 1994 Mar;40(3):367–71. [
PubMed: 8187301]
- 14.
Slavin RE, Walsh TJ, Pollack AD. Late generalized tuberculosis: a clinical pathologic analysis and comparison of 100 cases in the preantibiotic and antibiotic eras.
Medicine (Baltimore). 1980 Sep;59(5):352–66. [
PubMed: 7432152]
- 15.
Guttman P. Addison’s disease: A statistical analysis of 566 cases and a study of pathology. Arch Pathol. 1930;10:742–5.
- 16.
Sanford JP, Favour CB. The interrelationships between Addison's disease and active tuberculosis: a review of 125 cases of Addison's disease.
Ann Intern Med. 1956 Jul;45(1):56–72. [
PubMed: 13340568]
- 17.
Betterle C, Morlin L. Autoimmune Addison’s Disease.
Endocr Dev. 2011;20:171–72. [
PubMed: 21164269]
- 18.
Agarwal G, Bhatia E, Pandey R, Jain SK. Clinical profile and prognosis of Addison's disease in India.
Natl Med J India. 2001 Jan-Feb;14(1):23–5. [
PubMed: 11242694]
- 19.
Soule S. Addison's disease in Africa--a teaching hospital experience.
Clin Endocrinol (Oxf). 1999 Jan;50(1):115–20. [
PubMed: 10341864]
- 20.
- 21.
Alvarez S, McCabe WR. Extrapulmonary tuberculosis revisited: a review of experience at Boston City and other hospitals.
Med. 1984;63:25–55. [
PubMed: 6419006]
- 22.
Huang YC, Tang YL, Zhang XM, Zeng NL, Li R, Chen TW. Evaluation of primary adrenal insufficiency secondary to tuberculous adrenalitis with computed tomography and magnetic resonance imaging: Current status.
World J Radiol. 2015;28:336–42. [
PMC free article: PMC4620114] [
PubMed: 26516430]
- 23.
Maitra A. The endocrine system. In: Kumar V, Abbas AK, Aster AC, editors. Robbins and Cotran Pathologic Basis of Disease. Philadelphia; Elsevier. 9th edition. 2015. P. 1073-139.
- 24.
- 25.
Chhangani NP, Sharma P. Addison’s disease.
Indian Ped. 2003;40:904–5. [
PubMed: 14530557]
- 26.
Sharma S, Joshi R, Kalelkar R, Agrawal P. Tuberculous adrenal abscess presenting as adrenal insufficiency in a 4-year-old boy.
J Tropical Ped. 2018;0:1–4. [
PubMed: 30060233]
- 27.
Bancos I, Hahner S, Tomlinson J, Arlt W. Diagnosis and management of adrenal insufficiency.
Lancet Diabetes Endocrinol. 2015;3:216–26. [
PubMed: 25098712]
- 28.
Herndon J, Nadeau AM, Davidge-Pitts CJ, Young WF, Bancos I. Primary adrenal insufficiency due to bilateral infiltrative disease.
Endocrine. 2018;62:721–8. [
PubMed: 30178435]
- 29.
Erichsen MM, Lovas K, Skinningsrud B, Wolff AB, Undlien DE, Svatberg J, et al. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry.
J Clin Endocrinol Metab. 2009;94:4882–4890. [
PubMed: 19858318]
- 30.
Nair A, Jayakumari C, George GS, Jabbar PK, Das DV, Jessy SJ, Aneesh TS. Long acting porcine sequence ACTH in the diagnosis of adrenal insufficiency.
Eur J Endocrinol. 2019 Dec;181(6):639–645. [
PubMed: 31614334]
- 31.
Lam KY, Lo CY. A critical examination of adrenal tuberculosis and a 28-year autopsy experience of active tuberculosis.
Clin Endocrinol. 2001;54:633–9. [
PubMed: 11380494]
- 32.
Friedman F. The pathology of the adrenal gland in Addison’s disease with special reference to adrenocortical contraction. J name to be found 1948;42:181-200. [
PubMed: 18914213]
- 33.
Annear TD, Baker GP. Tuberculous Addison’s disease. A case apparently cured by chemotherapy.
Lancet. 1961;2:577–8. [
PubMed: 13683660]
- 34.
- 35.
- 36.
Bulbuloglu E, Ciralik H, Okur E, Ozdemir G, Ezberci F, Cetinkaya A. Tuberculosis of the thyroid gland: review of the literature.
World J Surg. 2006;30:149–55. [
PubMed: 16425087]
- 37.
- 38.
Das DK, Pant CS, Chachra KL, Gupta AK. Fine needle aspiration cytology diagnosis of tuberculous thyroiditis. A report of eight cases.
Acta Cytol. 1992;36:517–22. [
PubMed: 1636345]
- 39.
Mondal A, Patra DK. Efficacy of fine needle aspiration cytology in the diagnosis of tuberculosis of the thyroid gland: a study of 18 cases.
J Laryngol Otol. 1995;109:36–8. [
PubMed: 7876734]
- 40.
Gupta N, Sharma K, Barwad A, Sharma M, Rajwanshi A, Dutta P, et al. Thyroid tuberculosis – role of PCR in diagnosis of a rare entity.
Cytopathol. 2011;22:392–6. [
PubMed: 21118313]
- 41.
Ozekinci S, Mizrak B, Saruhan G, Senturk S. Histopathologic diagnosis of thyroid tuberculosis.
Thyroid. 2009;19:983–6. [
PubMed: 19678750]
- 42.
Akbulut S, Sogutcu N, Arikanoglu Z, Bakir S, Ulku A, Yagmur Y. Thyroid tuberculosis in Southeastern Turkey: is this the resurgence of a stubborn disease?
World J Surg. 2011;35:1847–52. [
PubMed: 21523497]
- 43.
- 44.
Luiz HV, Pereira BD, Silva TN, Veloza A, Matos A, Matos C, et al. Thyroid tuberculosis with abnormal thyroid function – case report and review of the literature.
Endocr Pract. 2013;19:e44–e49. [
PubMed: 23337150]
- 45.
Barnes P, Weatherstone R. Tuberculosis of the thyroid: two case reports.
Br J Dis Chest. 1979;73:187–91. [
PubMed: 119548]
- 46.
Kang BC, Lee SW, Shim SS, Choi HY, Baek SY, Cheon YJ. US.
Clin Imaging. 2000;24:283–6. and CT findings of tuberculosis of the thyroid: three case reports. [
PubMed: 11331157]
- 47.
Madhusudhan KS, Seith A, Khadgawat R, Das P, Mathur S. Tuberculosis of the thyroid gland: magnetic resonance imaging appearances.
Singapore Med J. 2009;50:e235–e238. [
PubMed: 19644607]
- 48.
El Malki HO, El Absi M, Mohsine R. …. Tuberculosis of the thyroid. Diagnosis and treatment.
Ann Chir. 2002;127:385–7. [
PubMed: 12094423]
- 49.
Chow CC, Mak TW, Chan CH, Cockram CS. Euthyroid sick syndrome in pulmonary tuberculosis before and after treatment.
Ann Clin Biochem. 1995;32:385–391. [
PubMed: 7486798]
- 50.
Christensen HR, Simonsen K, Hegedus L, Hansen BM, Dossing M, Kampmamn JP, et al. Influence of rifampicin on thyroid gland volume, thyroid hormones, and antipyrine metabolism.
Acta Endocrinol (Copenh). 1989;121:406–410. [
PubMed: 2800919]
- 51.
Kim HI, Kim TH, Kim H, Kim YN, Jang HW, Chung JH, et al. Effect of Rifampin on Thyroid Function Test in Patients on Levothyroxine Medication.
PLoS ONE. 2017;12(1):e0169775. [
PMC free article: PMC5231266] [
PubMed: 28081173]
- 52.
Munkner T. Studies on goiter due to para-aminosalicylic acid.
Scand J Respir Dis. 1969;50(3):212–26. [
PubMed: 4186670]
- 53.
Chhabra N, Gupta N, Aseri M L, Mathur SK, Dixit R. Analysis of thyroid function tests in patients of multidrug resistance tuberculosis undergoing treatment.
J Pharmacol Pharmacother. 2011;2:282–5. [
PMC free article: PMC3198526] [
PubMed: 22025859]
- 54.
Munivenkatappa S, Anil S, Naik B, et al. Drug-Induced Hypothyroidism during Anti-Tuberculosis Treatment of Multidrug-Resistant Tuberculosis: Notes from the Field.
Journal of Tuberculosis Research. 2016 Sep;4(3):105–110. [
PMC free article: PMC5007858] [
PubMed: 27595122]
- 55.
- 56.
Kirshbaum JD, Levy HA. Tuberculoma of hypophysis with insufficiency of anterior lobe: a clinical and pathological study of two cases. Arch Intern Med. 1941;68:1095–104.
- 57.
- 58.
Sunil K, Menon R, Goel N, Sanghvi D, Bandgar T, Joshi SR, et al. Pituitary tuberculosis.
J Assoc Physicians India. 2007;55:453–6. [
PubMed: 17879504]
- 59.
Dutta P, Bhansali A, Singh P, Bhat MH. Suprasellar tubercular abscess presenting as panhypopituitarism: a common lesion in an uncommon site with a brief review of literature.
Pituitary. 2006;6:73–7. [
PubMed: 16703412]
- 60.
Sharma MC, Arora R, Mahapatra AK, Sarat-Chandra P, Gaikwad SB, Sarkar C. Intrasellar tuberculoma – an enigmatic pituitary infection: a series of 18 cases.
Clin Neurol Neurosurg. 2000;102:72–7. [
PubMed: 10817892]
- 61.
- 62.
- 63.
Srisukh S, Tanpaibule T, Kiertiburanakul S, Boongird A, Wattanatranon D, Panyaping T, et al. Pituitary tuberculoma: a consideration in the differential diagnosis in a patient manifesting with pituitary apoplexy-like syndrome.
ID Cases. 2016;5:63–6. [
PMC free article: PMC4976610] [
PubMed: 27516966]
- 64.
- 65.
- 66.
Saini KS, Patel AL, Shaikh WA, Magar LN, Pungaonkar SA. Magnetic resonance spectroscopy in pituitary tuberculoma.
Singap Med J. 2007;48:783–6. [
PubMed: 17657390]
- 67.
Doknic M, Miljic D, Pekic S, Stojanovic M, Savic D, Manojlovic-Gacic E, et al. Single center study of 53 consecutive patients with pituitary stalk lesions.
Pituitary. 2018 [
PubMed: 30276501] [
CrossRef]
- 68.
Agrawal VM, Giri PJ. Tuberculosis: a common infection with rare presentation, isolated sellar tuberculoma with panhypopituitarism.
J Neurosci Rural Pract. 2019;10:327–30. [
PMC free article: PMC6454932] [
PubMed: 31001028]
- 69.
Antony G, Dasgupta R, Chacko G, Thomas N. Pituitary tuberculoma with subsequent drug-resistant tuberculous lymphadenopathy: an uncommon presentation of a common disease.
BMJ Case Rep. 2017 [
PMC free article: PMC5307273] [
PubMed: 28183710]
- 70.
Dhanwal DK, Vyas A, Sharma A, Saxena A. Hypothalamic pituitary abnormalities in tubercular meningitis at the time of diagnosis.
Pituitary. 2010;13:304–10. [
PubMed: 20495961]
- 71.
Mohammed H, Goyal MK, Dutta P, Sharma K, Modi M, et al. Hypothalamic and pituitary dysfunction is common in tubercular meningitis: A prospective study from a tertiary care center in Northern India.
J Neurol Sci. 2018 Dec 15;395:153–158. [
PubMed: 30321796]
- 72.
More A, Verma R, Garg RK, et al. A study of neuroendocrine dysfunction in patients of tuberculous meningitis.
J Neurol Sci. 2017 Aug 15;379:198–206. [
PubMed: 28716240]
- 73.
Lam KS, Shamm MM, Tam SC, Ng MM, Ma HT. Hypopituitarism after tuberculous meningitis in childhood.
Ann Intern Med. 1993;118:701–6. [
PubMed: 8460856]
- 74.
Chung DK, Hubbard WW. Hyponatremia in untreated active pulmonary tuberculosis.
Am Rev Respir Dis. 1969;99:595–597. [
PubMed: 5767592]
- 75.
Morris CD, Bird AR, Nell H. The haematological and biochemical changes in severe pulmonary tuberculosis.
Q J Med. 1989;73:1151–1159. [
PubMed: 2616737]
- 76.
Jonaidi Jafari N, Izadi M, Sarrafzadeh F, Heidari A, Ranjbar R, Saburi A. Hyponatremia due to pulmonary tuberculosis: review of 200 cases.
Nephrourol Mon. 2013;5:687–691. [
PMC free article: PMC3614323] [
PubMed: 23577332]
- 77.
Dash M, Sen RK, Behera BP, Sahu SS. Prevalence of hyponatremia in pulmonary tuberculosis. Int J Adv Med. 2020;7:63–6.
- 78.
Khan K, Rasool N, Mustafa F, Tariq R. Hyponatremia Due to Pulmonary Tuberculosis in Indian Population. Int J Sci Stud. 2017;5(5):98–101.
- 79.
Chaya BE, Rajesh KN, Mohan K, Mahesh DM. Hyponatremia in tuberculosis: Focus on brain instead of adrenals.
Neurol India. 2018;66:1515–6. [
PubMed: 30233041]
- 80.
Weiss H, Katz S. Hyponatremia resulting from apparently inappropriate secretion of antidiuretic hormone in patients with pulmonary tuberculosis.
Am Rev Respir Dis. 1965 Oct;92(4):609–16. [
PubMed: 5833838]
- 81.
Anderson RJ, Pluss RG, Berns AS, Jackson JT, Arnold PE, Schrier RW, McDonald KE. Mechanism of effect of hypoxia on renal water excretion.
J Clin Invest. 1978;62:769–777. [
PMC free article: PMC371828] [
PubMed: 701476]
- 82.
Hill AR, Uribarri J, Mann J, Berl T. Altered water metabolism in tuberculosis: role of vasopressin.
Am J Med. 1990;88:357–364. [
PubMed: 2327423]
- 83.
Lee P, Ho KK. Hyponatremia in pulmonary TB: evidence of ectopic antidiuretic hormone production.
Chest. 2010;137:207–208. [
PubMed: 20051406]
- 84.
Roca B, Tornador N, Tornador E. Presentation and outcome of tuberculous meningitis in adults in the province of Castellon, Spain: a retrospective study.
Epidemiol Infect. 2008;136:1455–1462. [
PMC free article: PMC2870755] [
PubMed: 18205976]
- 85.
Misra UK, Kalita J, Bhoi SK, Singh RK. A study of hyponatremia in tuberculous meningitis.
J Neurol Sci. 2016 Aug 15;367:152–7. [
PubMed: 27423581]
- 86.
Misra UK, Kalita J., Tuberculous Meningitis International Research Consortium. Mechanism, spectrum, consequences and management of hyponatremia in tuberculous meningitis.
Wellcome Open Research. 2019;4:189. [version 1; peer review: 2 approved] https://doi.org(accessed November 2020) [
PMC free article: PMC7372311] [
PubMed: 32734004] [
CrossRef]
- 87.
Cotton MF, Donald PR, Schoeman JF, Aalbers C, Van Zyl LE, Lombard C. Plasma arginine vasopressin and the syndrome of inappropriate antidiuretic hormone secretion in tuberculous meningitis.
Pediatr Infect Dis J. 1991;10:837–842. [
PubMed: 1844394]
- 88.
- 89.
Singh BS, Patwari AK, Deb M. Serum sodium and osmolal changes in tuberculous meningitis.
Indian Pediatr. 1994;31(11):1345–50. [
PubMed: 7896331]
- 90.
Cotton MF, Donald PR, Schoeman JF, Van Zyl LE, Aalbers C, Lombard CJ. Raised intracranial pressure, the syndrome of inappropriate antidiuretic hormone secretion, and arginine vasopressin in tuberculous meningitis.
Childs Nerv Syst. 1993;9:10–15. [
PubMed: 8481936]
- 91.
Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage.
Lancet. 1997;349(9047):245–59. [
PubMed: 9014912]
- 92.
Lenhard T, Külkens S, Schwab S. Cerebral salt-wasting syndrome in a patient with neuroleptic malignant syndrome.
Arch Neurol. 2007;64(1):122–25. [
PubMed: 17210819]
- 93.
Misra UK, Kalita J, Kumar M. Safety and Efficacy of Fludrocortisone in theTreatment of Cerebral Salt Wasting in Patients with Tuberculous Meningitis: A Randomized Clinical Trial.
JAMA Neurol. 2018;75(11):1383–91. [
PMC free article: PMC6248117] [
PubMed: 30105362]
- 94.
Jacobs TP, Bilezikian JP. Clinical review: Rare causes of hypercalcemia.
J Clin Endocrinol Metab. 2005 Nov;90(11):6316–22. [
PubMed: 16131579]
- 95.
Shek CC, Natkunam A, Tsang V, Cockram CS, Swaminathan R. Incidence, causes and mechanism of hypercalcaemia in a hospital population in Hong Kong.
Q J Med. 1990 Dec;77(284):1277–85. [
PubMed: 2290921]
- 96.
- 97.
Lind L, Ljunghall S. Hypercalcemia in pulmonary tuberculosis.
Ups J Med Sci. 1990;95(2):157–60. [
PubMed: 2075643]
- 98.
Liam CK, Lim KH, Srinivas P, Poi PJ. Hypercalcaemia in patients with newly diagnosed tuberculosis in Malaysia.
Int J Tuberc Lung Dis. 1998 Oct;2(10):818–23. [
PubMed: 9783529]
- 99.
Kitrou MP, Phytou-Pallikari A, Tzannes SE, Virvidakis K, Mountokalakis TD. Hypercalcemia in active pulmonary tuberculosis.
Ann Intern Med. 1982 Feb;96(2):255. [
PubMed: 7059082]
- 100.
Roussos A, Lagogianni I, Gonis A, Ilias I, Kazi D, Patsopoulos D, Philippou N. Hypercalcaemia in Greek patients with tuberculosis before the initiation of anti-tuberculosis treatment.
Respir Med. 2001 Mar;95(3):187–90. [
PubMed: 11266235]
- 101.
- 102.
A controlled trial of six months chemotherapy in pulmonary tuberculosis. First Report: results during chemotherapy. British Thoracic Association.
Br J Dis Chest. 1981 Apr;75(2):141–53. [
PubMed: 7023526]
- 103.
Keleştimur F, Güven M, Ozesmi M, Paşaoğlu H. Does tuberculosis really cause hypercalcemia?
J Endocrinol Invest. 1996 Nov;19(10):678–81. [
PubMed: 9007699]
- 104.
Fuss M, Karmali R, Pepersack T, Bergans A, Dierckx P, Prigogine T, Bergmann P, Corvilain J. Are tuberculous patients at a great risk from hypercalcemia?
Q J Med. 1988 Nov;69(259):869–78. [
PubMed: 3271334]
- 105.
Chan TY. Differences in vitamin D status and calcium intake: possible explanations for the regional variations in the prevalence of hypercalcemia in tuberculosis.
Calcif Tissue Int. 1997 Jan;60(1):91–3. [
PubMed: 9030487]
- 106.
John SM, Sagar S, Aparna JK, Joy S, Mishra AK. Risk factors for hypercalcemia in patients with tuberculosis.
Int J Mycobacteriol. 2020;9:7–11. [
PubMed: 32474481]
- 107.
Abbasi AA, Chemplavil JK, Farah S, Muller BF, Arnstein AR. Hypercalcemia in active pulmonary tuberculosis.
Ann Intern Med. 1979 Mar;90(3):324–8. [
PubMed: 426400]
- 108.
Felsenfeld AJ, Drezner MK, Llach F. Hypercalcemia and elevated calcitriol in a maintenance dialysis patient with tuberculosis.
Arch Intern Med. 1986 Oct;146(10):1941–5. [
PubMed: 3767540]
- 109.
Gkonos PJ, London R, Hendler ED. Hypercalcemia and elevated 1,25-dihydroxyvitamin D levels in a patient with end-stage renal disease and active tuberculosis.
N Engl J Med. 1984 Dec 27;311(26):1683–5. [
PubMed: 6548798]
- 110.
- 111.
Cadranel J, Garabedian M, Milleron B, Guillozo H, Akoun G, Hance AJ. 1,25(OH)2D2 production by T lymphocytes and alveolar macrophages recovered by lavage from normocalcemic patients with tuberculosis.
J Clin Invest. 1990 May;85(5):1588–93. [
PMC free article: PMC296610] [
PubMed: 2159024]
- 112.
Facchini L, Venturini E, Galli L, de Martino M, Chiappini E. Vitamin D and tuberculosis: a review on a hot topic.
J Chemother. 2015;27:128–138. [
PubMed: 26058744]
- 113.
Rockett KA, Brookes R, Udalova I, Vidal V, Hill AV, Kwiatkowski D. 1,25-Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage like cell line.
Infect Immun. 1998;66:5314–5321. [
PMC free article: PMC108664] [
PubMed: 9784538]
- 114.
Coussens AK, Wilkinson RJ, Hanifa Y, et al. Vitamin D accelerates resolution of inflammatory responses during tuberculosis treatment.
Proc Natl Acad Sci U S A. 2012;109:15449–15454. [
PMC free article: PMC3458393] [
PubMed: 22949664]
- 115.
Sinclair D, Abba K, Grobler L, Sudarsanam TD. Nutritional supplements for people being treated for active tuberculosis.
Cochrane Database Syst Rev. 2011 Nov 9;(11):CD006086. [
PubMed: 22071828]
- 116.
Brodie MJ, Boobis AR, Hillyard CJ, et al. Effect of Isoniazid on vitamin D metabolism and hepatic monooxygenase activity.
Clin Pharmacol Ther. 1981;30:363–370. [
PubMed: 7273600]
- 117.
Brodie MJ, Boobis AR, Dollery CT, et al. Rifampicin and vitamin D metabolism.
Clin Pharmacol Ther. 1980;27:810–814. [
PubMed: 7379450]